

Assessing the clinical utility of cancer genomic and proteomic data across tumor types
- PMID: 24952901
- PMCID: PMC4102885
- DOI: 10.1038/nbt.2940
Assessing the clinical utility of cancer genomic and proteomic data across tumor types
Abstract
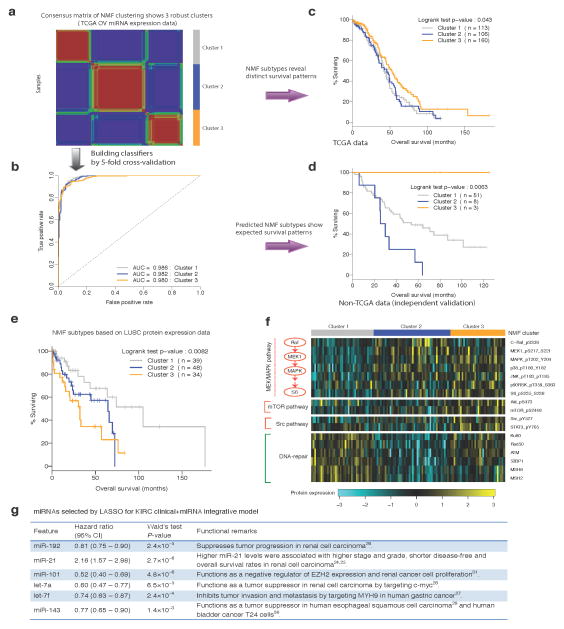
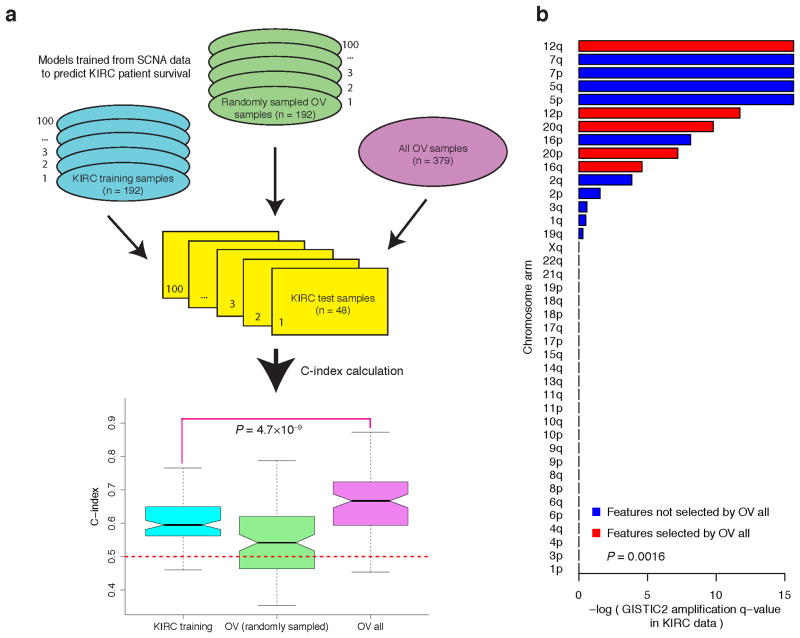
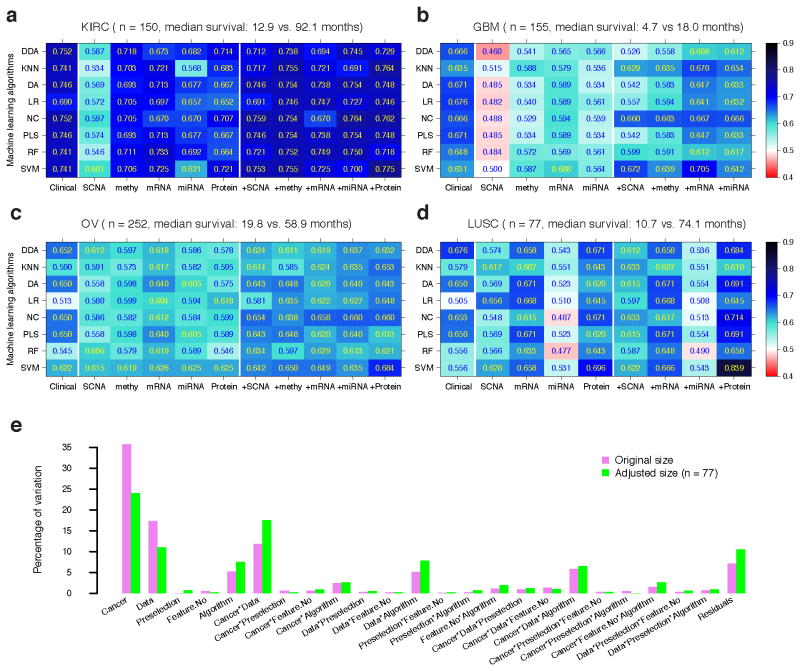
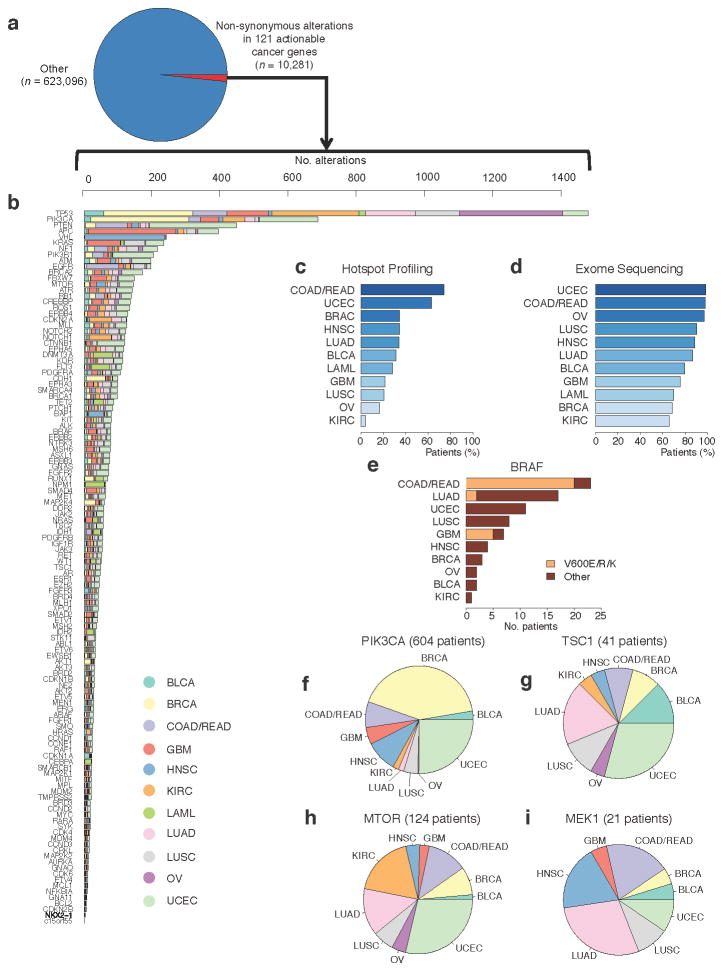
Molecular profiling of tumors promises to advance the clinical management of cancer, but the benefits of integrating molecular data with traditional clinical variables have not been systematically studied. Here we retrospectively predict patient survival using diverse molecular data (somatic copy-number alteration, DNA methylation and mRNA, microRNA and protein expression) from 953 samples of four cancer types from The Cancer Genome Atlas project. We find that incorporating molecular data with clinical variables yields statistically significantly improved predictions (FDR < 0.05) for three cancers but those quantitative gains were limited (2.2-23.9%). Additional analyses revealed little predictive power across tumor types except for one case. In clinically relevant genes, we identified 10,281 somatic alterations across 12 cancer types in 2,928 of 3,277 patients (89.4%), many of which would not be revealed in single-tumor analyses. Our study provides a starting point and resources, including an open-access model evaluation platform, for building reliable prognostic and therapeutic strategies that incorporate molecular data.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
