

Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database
- PMID: 24954049
- PMCID: PMC4090281
- DOI: 10.1016/j.cub.2014.05.044
Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database
Abstract
Background: Mass spectrometry-based proteomics enables the global identification and quantification of proteins and their posttranslational modifications in complex biological samples. However, proteomic analysis requires a complete and accurate reference set of proteins and is therefore largely restricted to model organisms with sequenced genomes.
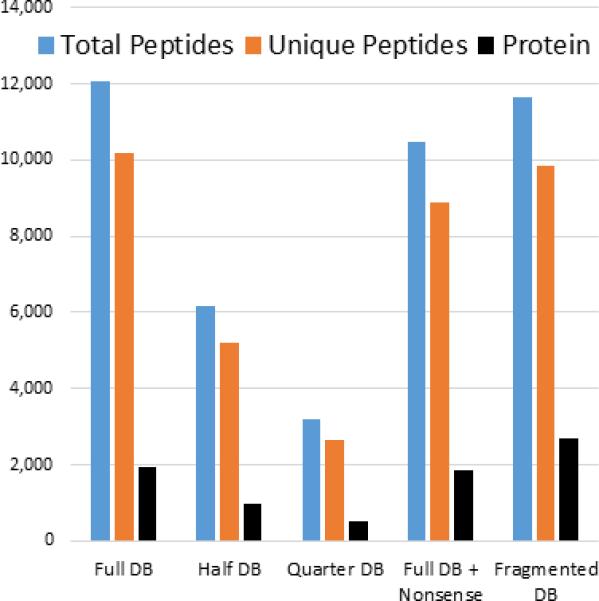
Results: Here, we demonstrate the feasibility of deep genome-free proteomics by using a reference proteome derived from heterogeneous mRNA data. We identify more than 11,000 proteins with 99% confidence from the unfertilized Xenopus laevis egg and estimate protein abundance with approximately 2-fold precision. Our reference database outperforms the provisional gene models based on genomic DNA sequencing and references generated by other methods. Surprisingly, we find that many proteins in the egg lack mRNA support and that many of these proteins are found in blood or liver, suggesting that they are taken up from the blood plasma, together with yolk, during oocyte growth and maturation, potentially contributing to early embryogenesis.
Conclusion: To facilitate proteomics in nonmodel organisms, we make our platform available as an online resource that converts heterogeneous mRNA data into a protein reference set. Thus, we demonstrate the feasibility and power of genome-free proteomics while shedding new light on embryogenesis in vertebrates.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures




References
-
- Kragl M, Knapp D, Nacu E, Khattak S, Maden M, Epperlein HH, Tanaka EM. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature. 2009;460:60–65. - PubMed
-
- di Prisco G, Cocca E, Parker S, Detrich H. Tracking the evolutionary loss of hemoglobin expression by the white-blooded Antarctic icefishes. Gene. 2002;295:185–191. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources