

Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth
- PMID: 24954902
- PMCID: PMC4376326
- DOI: 10.1016/j.molcel.2014.05.020
Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth
Abstract
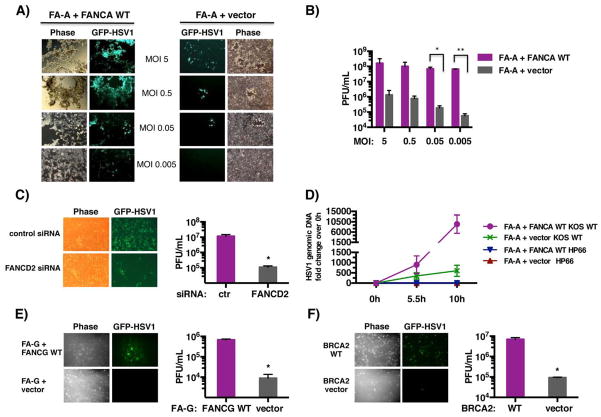
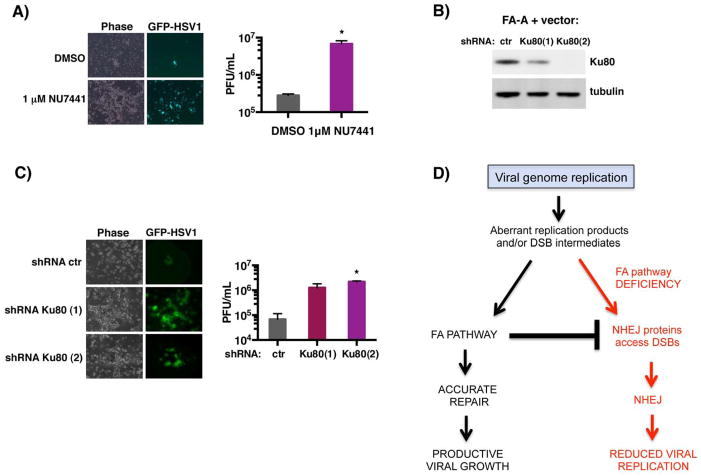
DNA damage associated with viral DNA synthesis can result in double-strand breaks that threaten genome integrity and must be repaired. Here, we establish that the cellular Fanconi anemia (FA) genomic stability pathway is exploited by herpes simplex virus 1 (HSV-1) to promote viral DNA synthesis and enable its productive growth. Potent FA pathway activation in HSV-1-infected cells resulted in monoubiquitination of FA effector proteins FANCI and FANCD2 (FANCI-D2) and required the viral DNA polymerase. FANCD2 relocalized to viral replication compartments, and FANCI-D2 interacted with a multisubunit complex containing the virus-encoded single-stranded DNA-binding protein ICP8. Significantly, whereas HSV-1 productive growth was impaired in monoubiquitination-defective FA cells, this restriction was partially surmounted by antagonizing the DNA-dependent protein kinase (DNA-PK), a critical enzyme required for nonhomologous end-joining (NHEJ). This identifies the FA-pathway as a cellular factor required for herpesvirus productive growth and suggests that FA-mediated suppression of NHEJ is a fundamental step in the viral life cycle.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays.Elife. 2020 Mar 13;9:e54128. doi: 10.7554/eLife.54128. Elife. 2020. PMID: 32167469 Free PMC article.
-
The ubiquitination machinery of the Fanconi Anemia DNA repair pathway.Prog Biophys Mol Biol. 2021 Aug;163:5-13. doi: 10.1016/j.pbiomolbio.2020.09.009. Epub 2020 Oct 12. Prog Biophys Mol Biol. 2021. PMID: 33058944 Review.
-
Regulation of the Fanconi Anemia DNA Repair Pathway by Phosphorylation and Monoubiquitination.Genes (Basel). 2021 Nov 5;12(11):1763. doi: 10.3390/genes12111763. Genes (Basel). 2021. PMID: 34828369 Free PMC article. Review.
-
[The role of the Fanconi anemia pathway in DNA repair and maintenance of genome stability].Postepy Hig Med Dosw (Online). 2014 May 8;68:459-72. doi: 10.5604/17322693.1101567. Postepy Hig Med Dosw (Online). 2014. PMID: 24864098 Review. Polish.
-
HIV-1 exploits the Fanconi anemia pathway for viral DNA integration.Cell Rep. 2022 May 24;39(8):110840. doi: 10.1016/j.celrep.2022.110840. Cell Rep. 2022. PMID: 35613597 Free PMC article.
Cited by
-
Identifying Protein Interactions with Viral DNA Genomes during Virus Infection.Viruses. 2024 May 25;16(6):845. doi: 10.3390/v16060845. Viruses. 2024. PMID: 38932138 Free PMC article. Review.
-
G2/M cell cycle arrest correlates with primate lentiviral Vpr interaction with the SLX4 complex.J Virol. 2015 Jan;89(1):230-40. doi: 10.1128/JVI.02307-14. Epub 2014 Oct 15. J Virol. 2015. PMID: 25320300 Free PMC article.
-
NEDDylation is essential for Kaposi's sarcoma-associated herpesvirus latency and lytic reactivation and represents a novel anti-KSHV target.PLoS Pathog. 2015 Mar 20;11(3):e1004771. doi: 10.1371/journal.ppat.1004771. eCollection 2015 Mar. PLoS Pathog. 2015. PMID: 25794275 Free PMC article.
-
FANCD2 Binds Human Papillomavirus Genomes and Associates with a Distinct Set of DNA Repair Proteins to Regulate Viral Replication.mBio. 2017 Feb 14;8(1):e02340-16. doi: 10.1128/mBio.02340-16. mBio. 2017. PMID: 28196964 Free PMC article.
-
Suppression of non-homologous end joining does not rescue DNA repair defects in Fanconi anemia patient cells.Cell Cycle. 2020 Oct;19(19):2553-2561. doi: 10.1080/15384101.2020.1810394. Epub 2020 Aug 30. Cell Cycle. 2020. PMID: 32865112 Free PMC article.
References
-
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing non-homologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. - PubMed
-
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497–510. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
