

iDrug: a web-accessible and interactive drug discovery and design platform
- PMID: 24955134
- PMCID: PMC4046018
- DOI: 10.1186/1758-2946-6-28
iDrug: a web-accessible and interactive drug discovery and design platform
Abstract
Background: The progress in computer-aided drug design (CADD) approaches over the past decades accelerated the early-stage pharmaceutical research. Many powerful standalone tools for CADD have been developed in academia. As programs are developed by various research groups, a consistent user-friendly online graphical working environment, combining computational techniques such as pharmacophore mapping, similarity calculation, scoring, and target identification is needed.
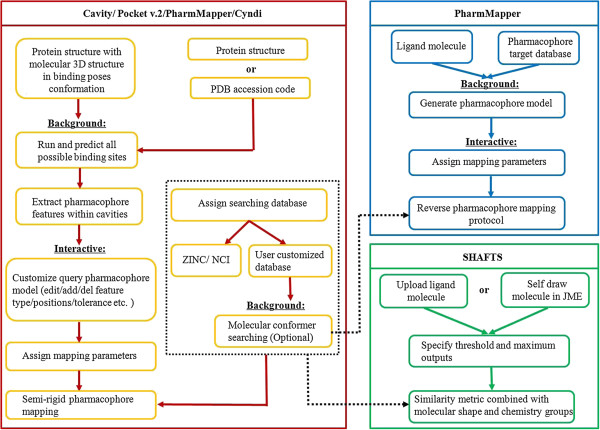
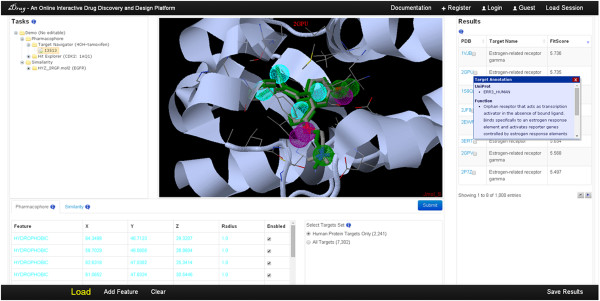
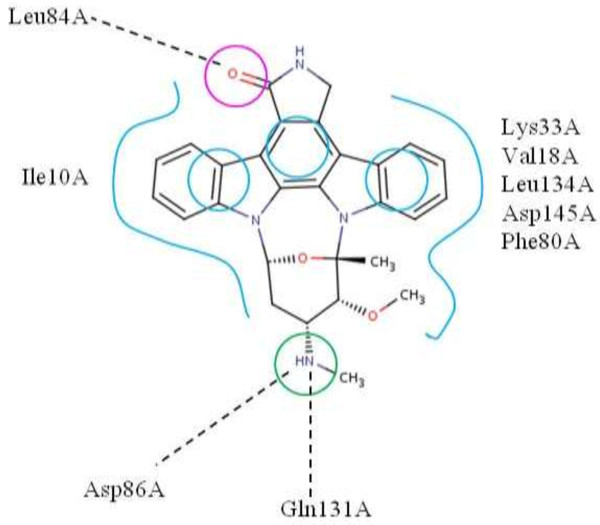
Results: We presented a versatile, user-friendly, and efficient online tool for computer-aided drug design based on pharmacophore and 3D molecular similarity searching. The web interface enables binding sites detection, virtual screening hits identification, and drug targets prediction in an interactive manner through a seamless interface to all adapted packages (e.g., Cavity, PocketV.2, PharmMapper, SHAFTS). Several commercially available compound databases for hit identification and a well-annotated pharmacophore database for drug targets prediction were integrated in iDrug as well. The web interface provides tools for real-time molecular building/editing, converting, displaying, and analyzing. All the customized configurations of the functional modules can be accessed through featured session files provided, which can be saved to the local disk and uploaded to resume or update the history work.
Conclusions: iDrug is easy to use, and provides a novel, fast and reliable tool for conducting drug design experiments. By using iDrug, various molecular design processing tasks can be submitted and visualized simply in one browser without installing locally any standalone modeling softwares. iDrug is accessible free of charge at http://lilab.ecust.edu.cn/idrug.
Keywords: 3D similarity calculation; Cavity detection; Online drug design platform; Pharmacophore search; Target prediction.
Figures
Similar articles
-
eSHAFTS: Integrated and graphical drug design software based on 3D molecular similarity.J Comput Chem. 2019 Mar 5;40(6):826-838. doi: 10.1002/jcc.25769. J Comput Chem. 2019. PMID: 30623477
-
PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database.Nucleic Acids Res. 2017 Jul 3;45(W1):W356-W360. doi: 10.1093/nar/gkx374. Nucleic Acids Res. 2017. PMID: 28472422 Free PMC article.
-
iDrug-Target: predicting the interactions between drug compounds and target proteins in cellular networking via benchmark dataset optimization approach.J Biomol Struct Dyn. 2015;33(10):2221-33. doi: 10.1080/07391102.2014.998710. Epub 2015 Jan 14. J Biomol Struct Dyn. 2015. PMID: 25513722
-
Open-Source Browser-Based Tools for Structure-Based Computer-Aided Drug Discovery.Molecules. 2022 Jul 20;27(14):4623. doi: 10.3390/molecules27144623. Molecules. 2022. PMID: 35889494 Free PMC article. Review.
-
Rational Drug Design of Antineoplastic Agents Using 3D-QSAR, Cheminformatic, and Virtual Screening Approaches.Curr Med Chem. 2019;26(21):3874-3889. doi: 10.2174/0929867324666170712115411. Curr Med Chem. 2019. PMID: 28707592 Review.
Cited by
-
Hydroxysafflor Yellow A Inhibits LPS-Induced NLRP3 Inflammasome Activation via Binding to Xanthine Oxidase in Mouse RAW264.7 Macrophages.Mediators Inflamm. 2016;2016:8172706. doi: 10.1155/2016/8172706. Epub 2016 Jun 28. Mediators Inflamm. 2016. PMID: 27433030 Free PMC article.
-
Mining Chemical Activity Status from High-Throughput Screening Assays.PLoS One. 2015 Dec 14;10(12):e0144426. doi: 10.1371/journal.pone.0144426. eCollection 2015. PLoS One. 2015. PMID: 26658480 Free PMC article.
-
Applying high-performance computing in drug discovery and molecular simulation.Natl Sci Rev. 2016 Mar;3(1):49-63. doi: 10.1093/nsr/nww003. Epub 2016 Jan 11. Natl Sci Rev. 2016. PMID: 32288960 Free PMC article.
-
DRABAL: novel method to mine large high-throughput screening assays using Bayesian active learning.J Cheminform. 2016 Nov 10;8:64. doi: 10.1186/s13321-016-0177-8. eCollection 2016. J Cheminform. 2016. PMID: 27895719 Free PMC article.
-
Using Big Data to Discover Diagnostics and Therapeutics for Gastrointestinal and Liver Diseases.Gastroenterology. 2017 Jan;152(1):53-67.e3. doi: 10.1053/j.gastro.2016.09.065. Epub 2016 Oct 20. Gastroenterology. 2017. PMID: 27773806 Free PMC article. Review.
References
-
- Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief Bioinform. 2009;10:579–591. - PubMed
-
- Jorgensen WL. The many roles of computation in drug discovery. Science. 2004;303:1813–1818. - PubMed
-
- Kellenberger E, Foata N, Rognan D. Ranking targets in structure-based virtual screening of three-dimensional protein libraries: methods and problems. J Chem Inf Model. 2008;48:1014–1025. - PubMed
-
- Nettles JH, Jenkins JL, Bender A, Deng Z, Davies JW, Glick M. Bridging chemical and biological space: “target fishing” using 2D and 3D molecular descriptors. J Med Chem. 2006;49:6802–6810. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
