

Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants
- PMID: 24956270
- PMCID: PMC4067418
- DOI: 10.1371/journal.pone.0100924
Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants
Abstract
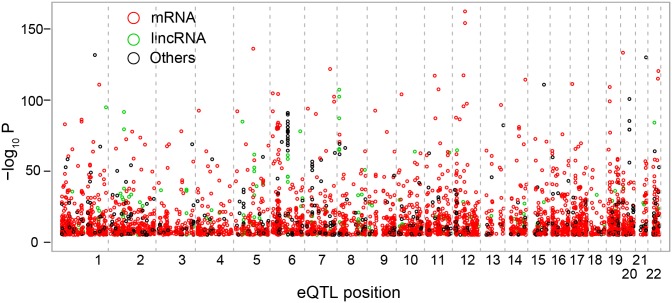
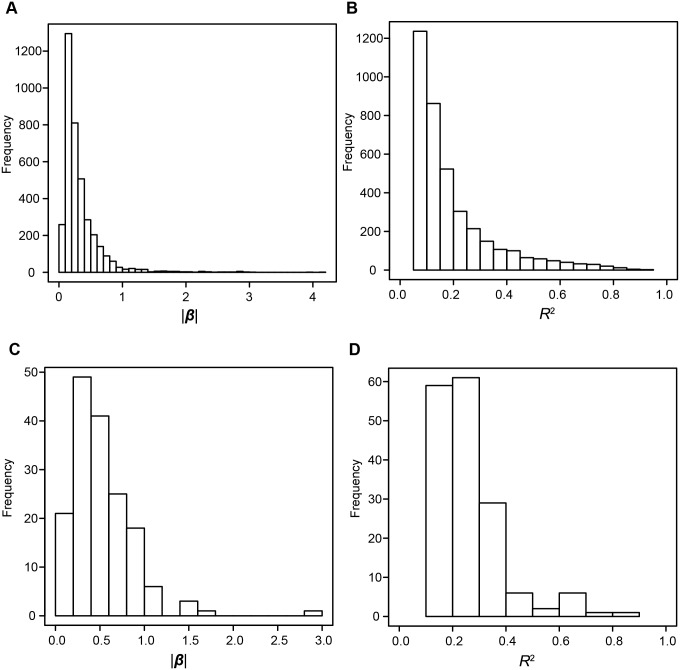
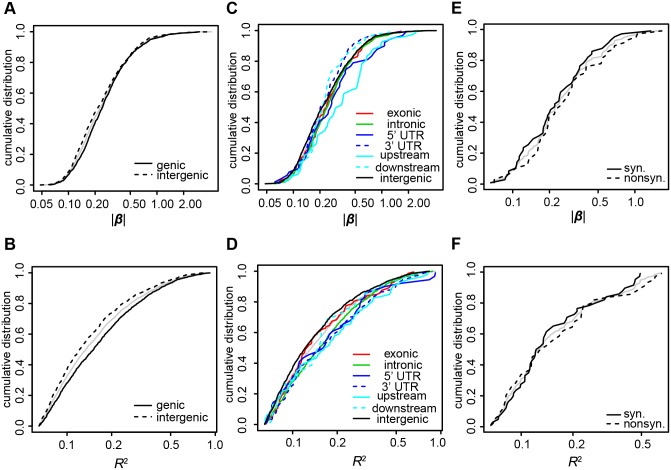
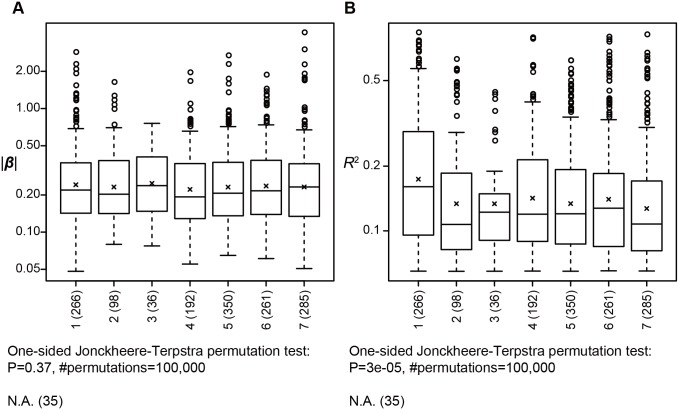
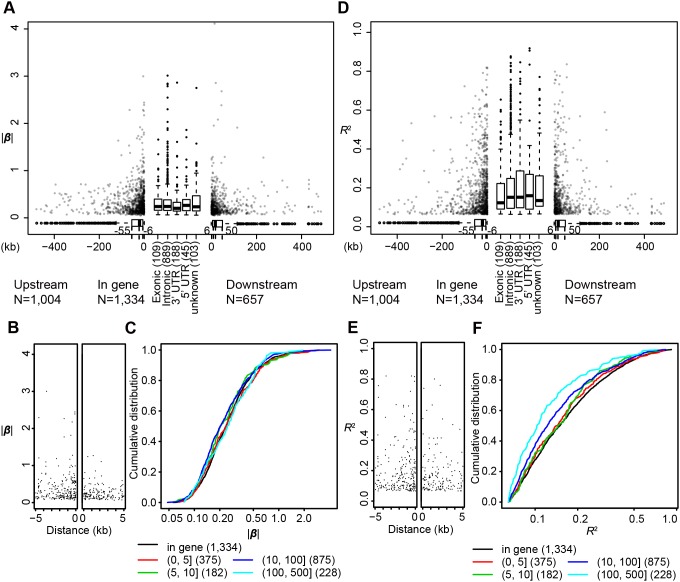
Profiles of sequence variants that influence gene transcription are very important for understanding mechanisms that affect phenotypic variation and disease susceptibility. Using genotypes at 1.4 million SNPs and a comprehensive transcriptional profile of 15,454 coding genes and 6,113 lincRNA genes obtained from peripheral blood cells of 298 Japanese individuals, we mapped expression quantitative trait loci (eQTLs). We identified 3,804 cis-eQTLs (within 500 kb from target genes) and 165 trans-eQTLs (>500 kb away or on different chromosomes). Cis-eQTLs were often located in transcribed or adjacent regions of genes; among these regions, 5' untranslated regions and 5' flanking regions had the largest effects. Epigenetic evidence for regulatory potential accumulated in public databases explained the magnitude of the effects of our eQTLs. Cis-eQTLs were often located near the respective target genes, if not within genes. Large effect sizes were observed with eQTLs near target genes, and effect sizes were obviously attenuated as the eQTL distance from the gene increased. Using a very stringent significance threshold, we identified 165 large-effect trans-eQTLs. We used our eQTL map to assess 8,069 disease-associated SNPs identified in 1,436 genome-wide association studies (GWAS). We identified genes that might be truly causative, but GWAS might have failed to identify for 148 out of the GWAS-identified SNPs; for example, TUFM (P = 3.3E-48) was identified for inflammatory bowel disease (early onset); ZFP90 (P = 4.4E-34) for ulcerative colitis; and IDUA (P = 2.2E-11) for Parkinson's disease. We identified four genes (P<2.0E-14) that might be related to three diseases and two hematological traits; each expression is regulated by trans-eQTLs on a different chromosome than the gene.
Conflict of interest statement
Figures
References
-
- Göring HHH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, et al. (2007) Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet 39: 1208–1216. - PubMed
-
- Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, et al. (2007) A genome-wide association study of global gene expression. Nat Genet 39: 1202–1207. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
