

Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease
- PMID: 24958103
- PMCID: PMC4135549
- DOI: 10.1128/MCB.00687-14
Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease
Abstract
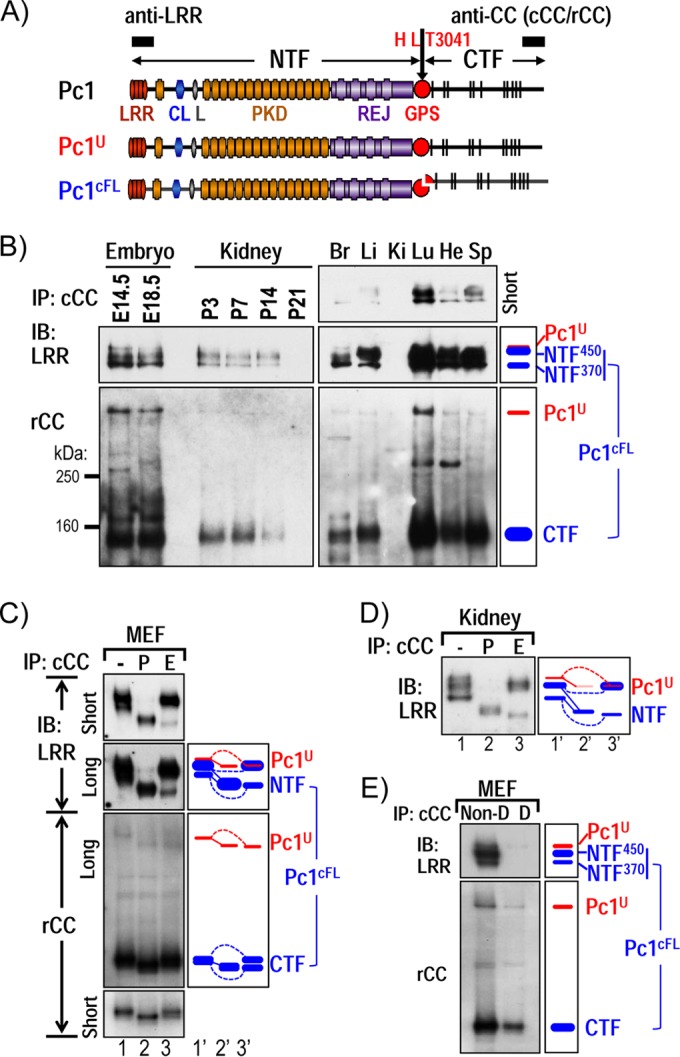
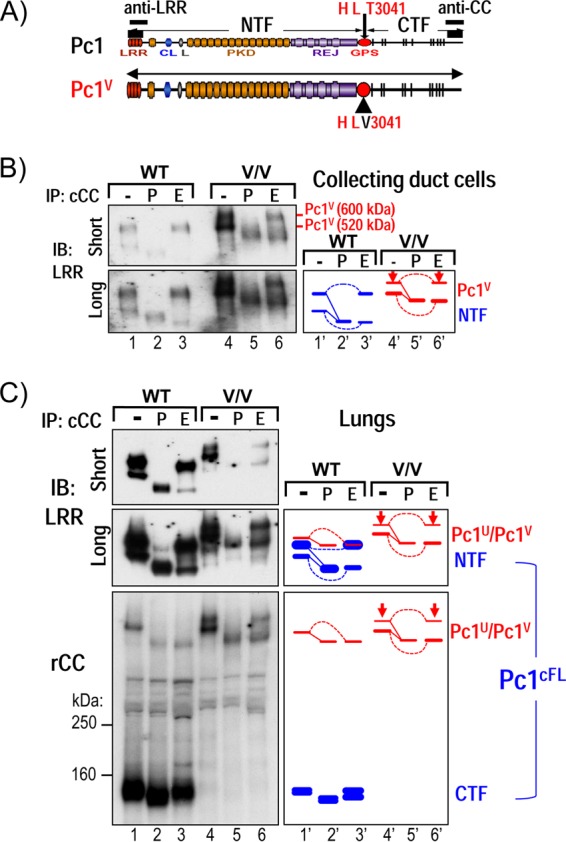
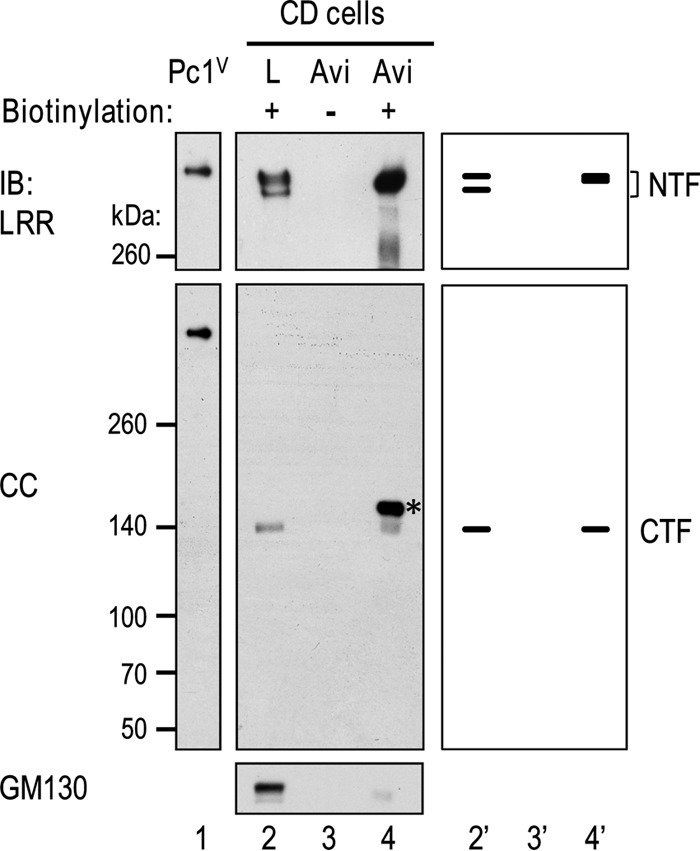
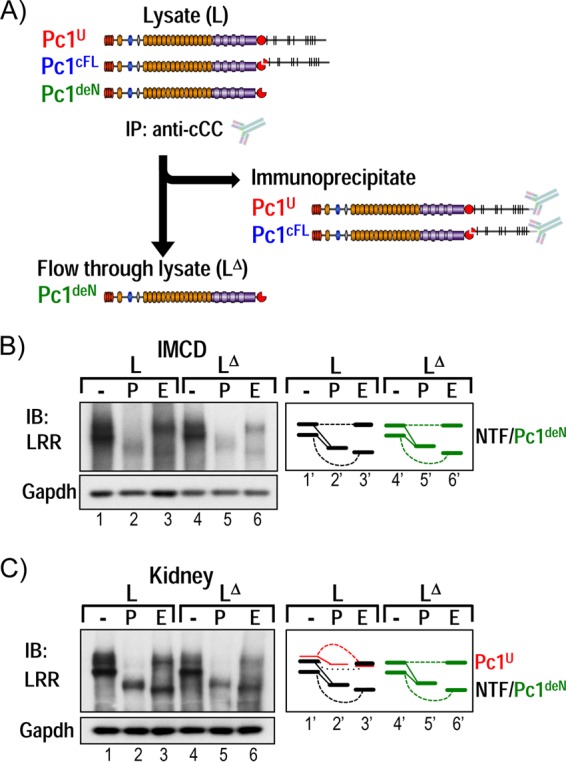
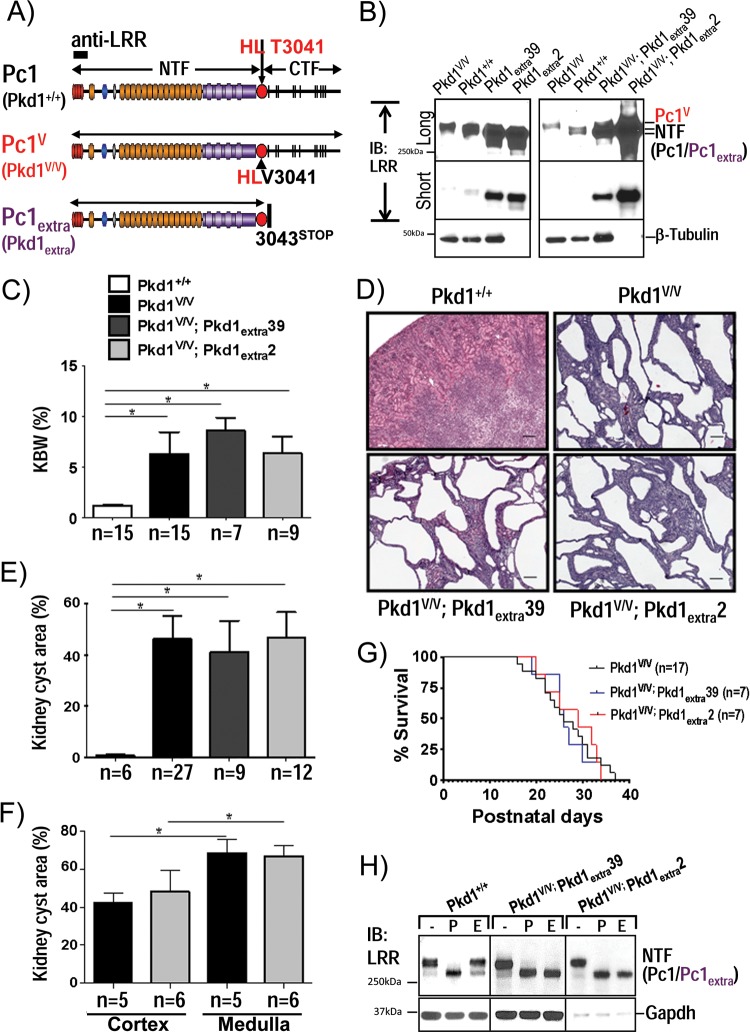
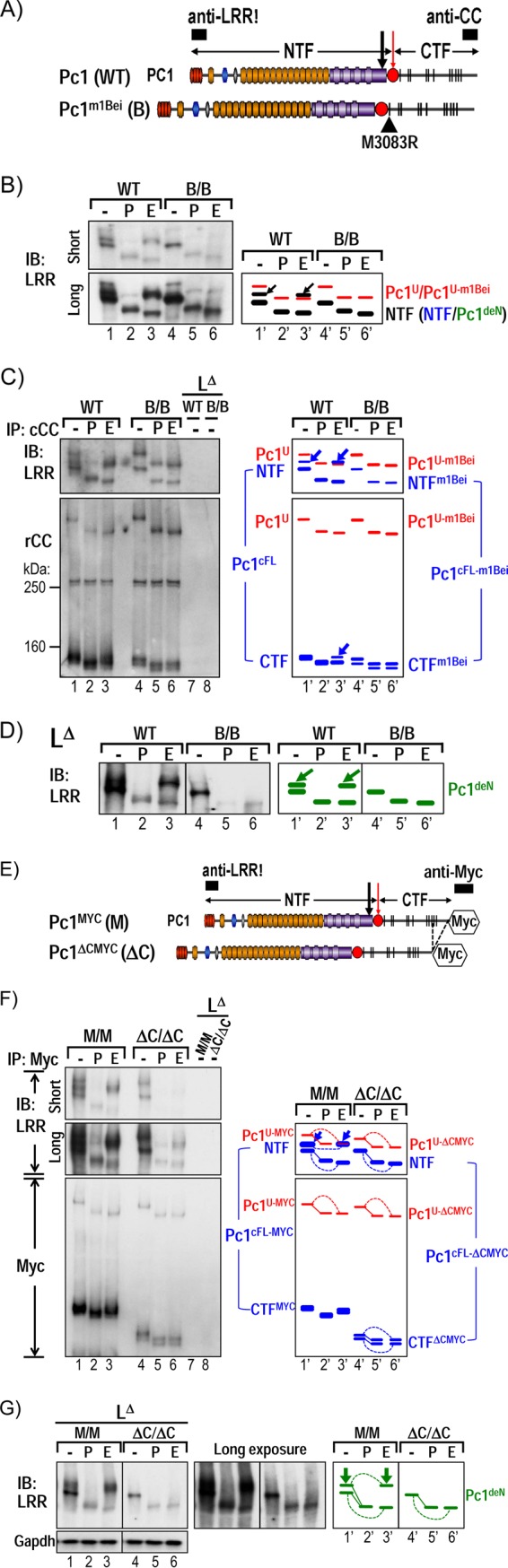
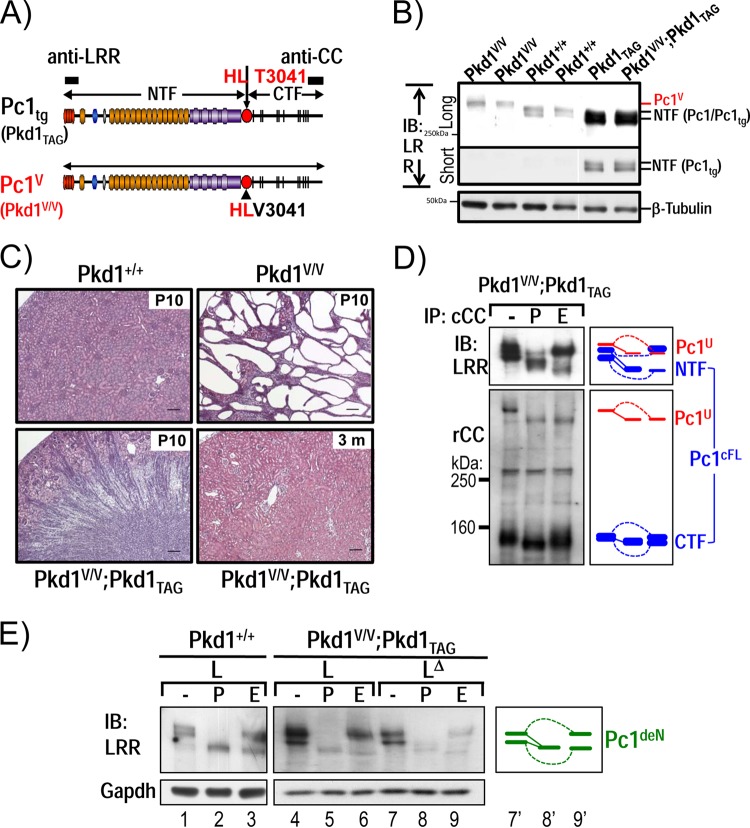
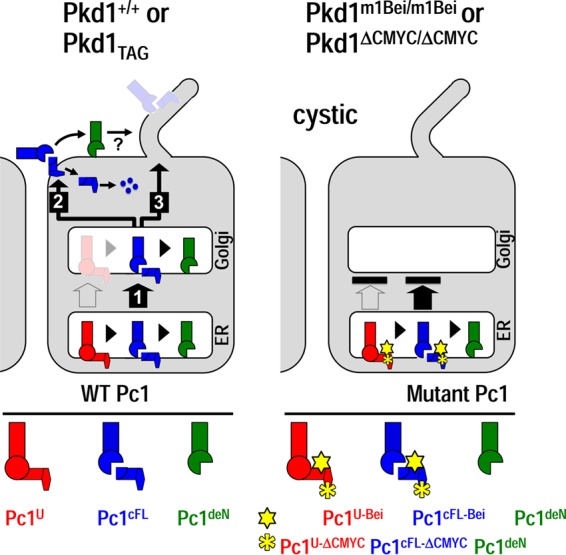
Polycystin-1 (Pc1) cleavage at the G protein-coupled receptor (GPCR) proteolytic site (GPS) is required for normal kidney morphology in humans and mice. We found a complex pattern of endogenous Pc1 forms by GPS cleavage. GPS cleavage generates not only the heterodimeric cleaved full-length Pc1 (Pc1(cFL)) in which the N-terminal fragment (NTF) remains noncovalently associated with the C-terminal fragment (CTF) but also a novel (Pc1) form (Pc1(deN)) in which NTF becomes detached from CTF. Uncleaved Pc1 (Pc1(U)) resides primarily in the endoplasmic reticulum (ER), whereas both Pc1(cFL) and Pc1(deN) traffic through the secretory pathway in vivo. GPS cleavage is not a prerequisite, however, for Pc1 trafficking in vivo. Importantly, Pc1(deN) is predominantly found at the plasma membrane of renal epithelial cells. By functional genetic complementation with five Pkd1 mouse models, we discovered that CTF plays a crucial role in Pc1(deN) trafficking. Our studies support GPS cleavage as a critical regulatory mechanism of Pc1 biogenesis and trafficking for proper kidney development and homeostasis.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Regulation of polycystin expression, maturation and trafficking.Cell Signal. 2020 Aug;72:109630. doi: 10.1016/j.cellsig.2020.109630. Epub 2020 Apr 8. Cell Signal. 2020. PMID: 32275942 Free PMC article. Review.
-
Polycystin-1 surface localization is stimulated by polycystin-2 and cleavage at the G protein-coupled receptor proteolytic site.Mol Biol Cell. 2010 Dec;21(24):4338-48. doi: 10.1091/mbc.E10-05-0407. Epub 2010 Oct 27. Mol Biol Cell. 2010. PMID: 20980620 Free PMC article.
-
Sec63 and Xbp1 regulate IRE1α activity and polycystic disease severity.J Clin Invest. 2015 May;125(5):1955-67. doi: 10.1172/JCI78863. Epub 2015 Apr 6. J Clin Invest. 2015. PMID: 25844898 Free PMC article.
-
Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1.PLoS One. 2010 Aug 23;5(8):e12305. doi: 10.1371/journal.pone.0012305. PLoS One. 2010. PMID: 20808796 Free PMC article.
-
The Role of G-protein Coupled Receptor Proteolytic Site (GPS) Cleavage in Polycystin-1 Biogenesis, Trafficking and Function.In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015 Nov. Chapter 11. In: Li X, editor. Polycystic Kidney Disease [Internet]. Brisbane (AU): Codon Publications; 2015 Nov. Chapter 11. PMID: 27512772 Free Books & Documents. Review.
Cited by
-
A scalable organoid model of human autosomal dominant polycystic kidney disease for disease mechanism and drug discovery.Cell Stem Cell. 2022 Jul 7;29(7):1083-1101.e7. doi: 10.1016/j.stem.2022.06.005. Cell Stem Cell. 2022. PMID: 35803227 Free PMC article.
-
Regulation of polycystin expression, maturation and trafficking.Cell Signal. 2020 Aug;72:109630. doi: 10.1016/j.cellsig.2020.109630. Epub 2020 Apr 8. Cell Signal. 2020. PMID: 32275942 Free PMC article. Review.
-
Cilia-Localized Counterregulatory Signals as Drivers of Renal Cystogenesis.Front Mol Biosci. 2022 Jun 23;9:936070. doi: 10.3389/fmolb.2022.936070. eCollection 2022. Front Mol Biosci. 2022. PMID: 35832738 Free PMC article. Review.
-
Long noncoding RNA Hoxb3os is dysregulated in autosomal dominant polycystic kidney disease and regulates mTOR signaling.J Biol Chem. 2018 Jun 15;293(24):9388-9398. doi: 10.1074/jbc.RA118.001723. Epub 2018 May 1. J Biol Chem. 2018. PMID: 29716997 Free PMC article.
-
Adhesion GPCRs as a paradigm for understanding polycystin-1 G protein regulation.Cell Signal. 2020 Aug;72:109637. doi: 10.1016/j.cellsig.2020.109637. Epub 2020 Apr 16. Cell Signal. 2020. PMID: 32305667 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous