

Hs3st3-modified heparan sulfate controls KIT+ progenitor expansion by regulating 3-O-sulfotransferases
- PMID: 24960693
- PMCID: PMC4105206
- DOI: 10.1016/j.devcel.2014.04.024
Hs3st3-modified heparan sulfate controls KIT+ progenitor expansion by regulating 3-O-sulfotransferases
Abstract
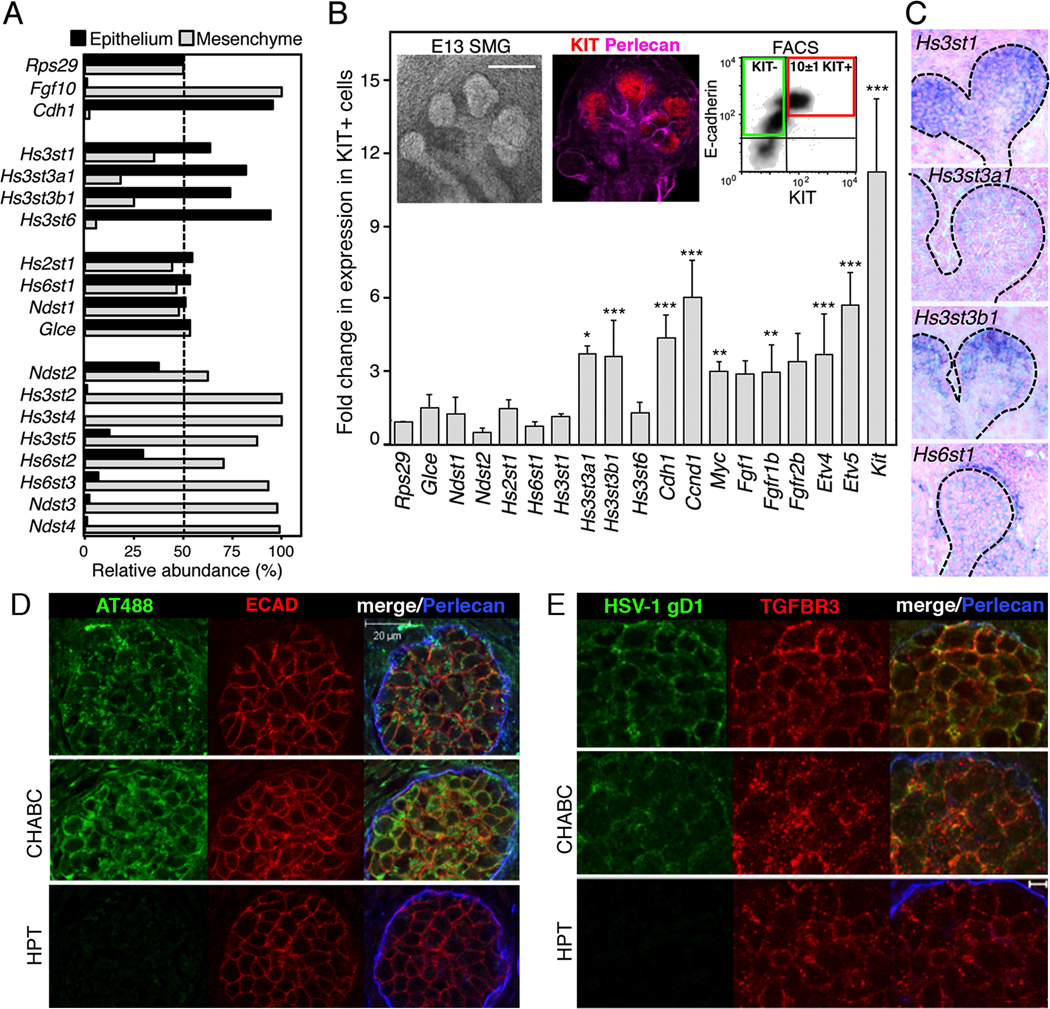
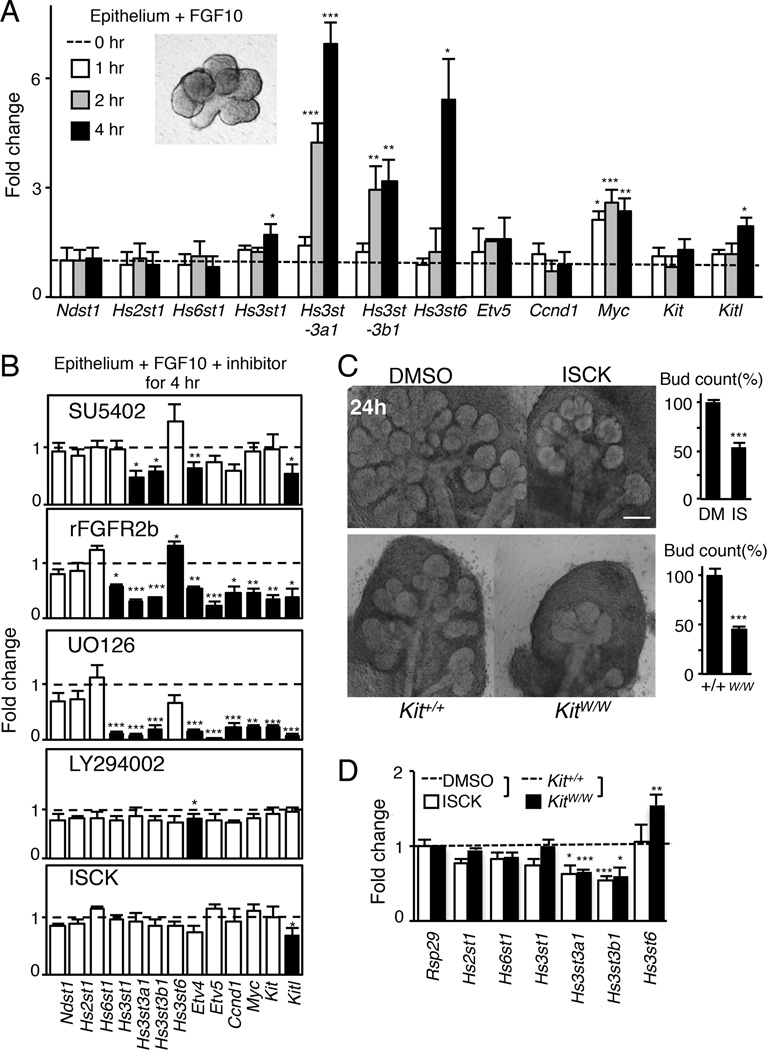
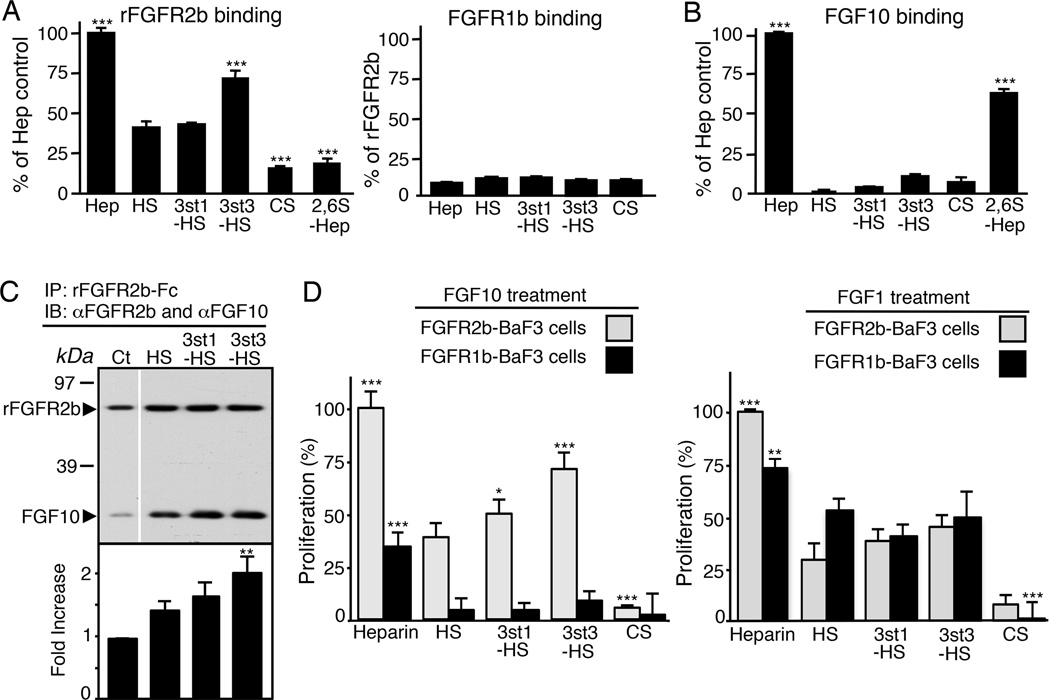
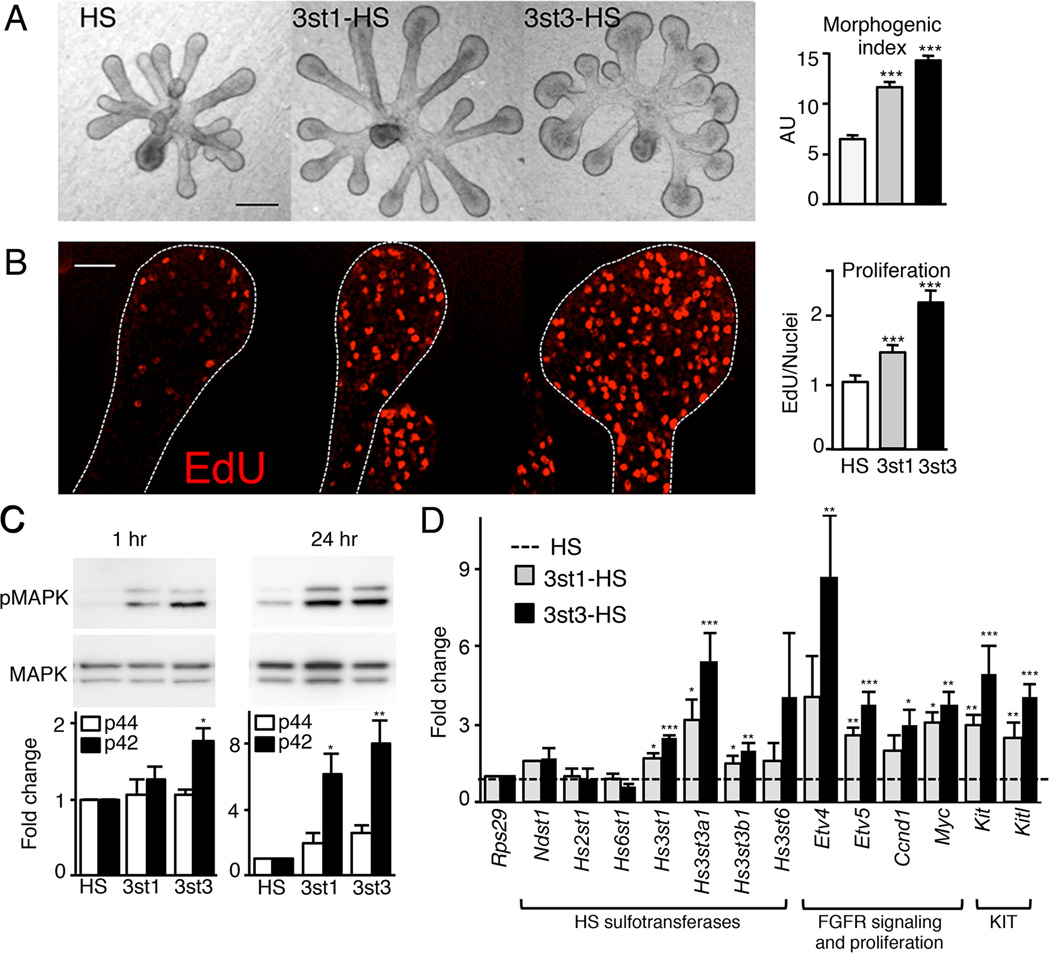
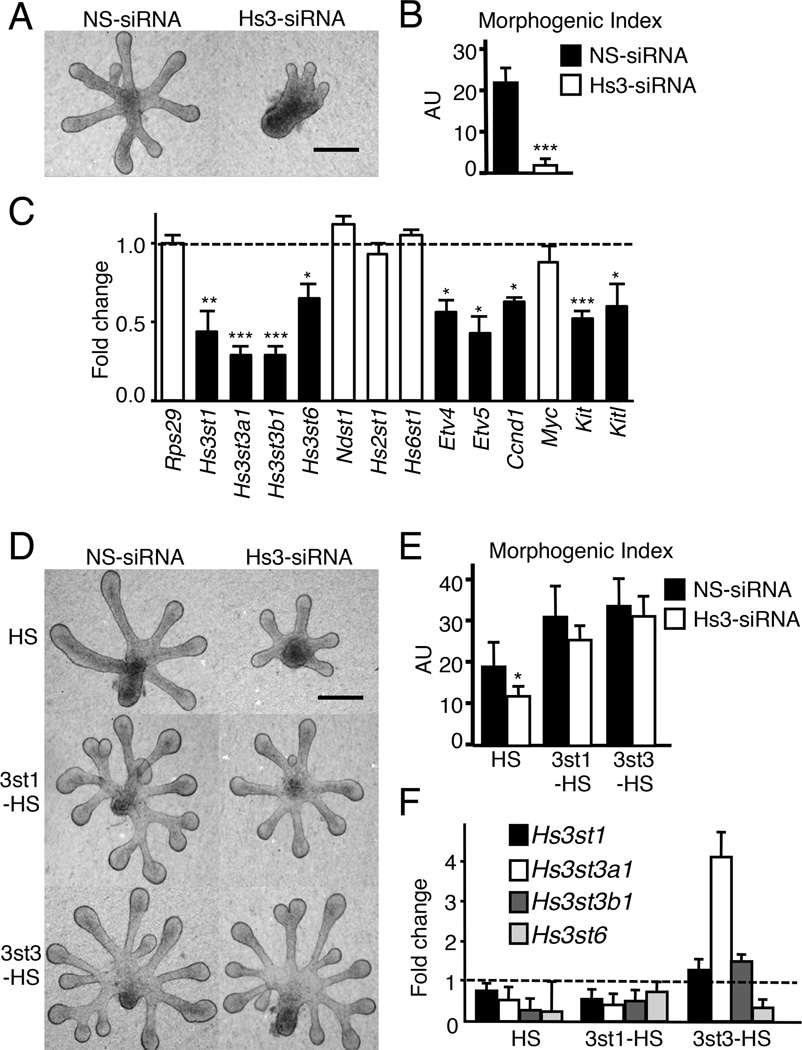
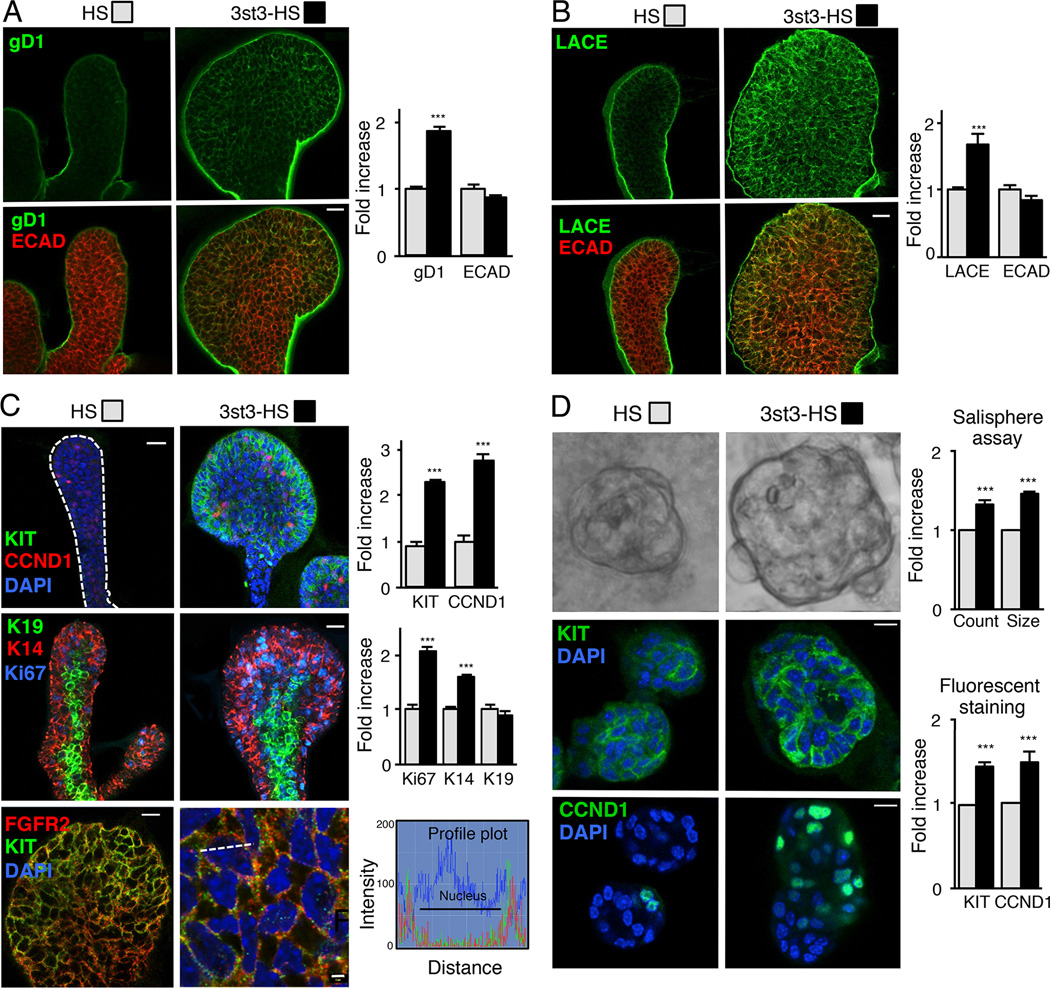
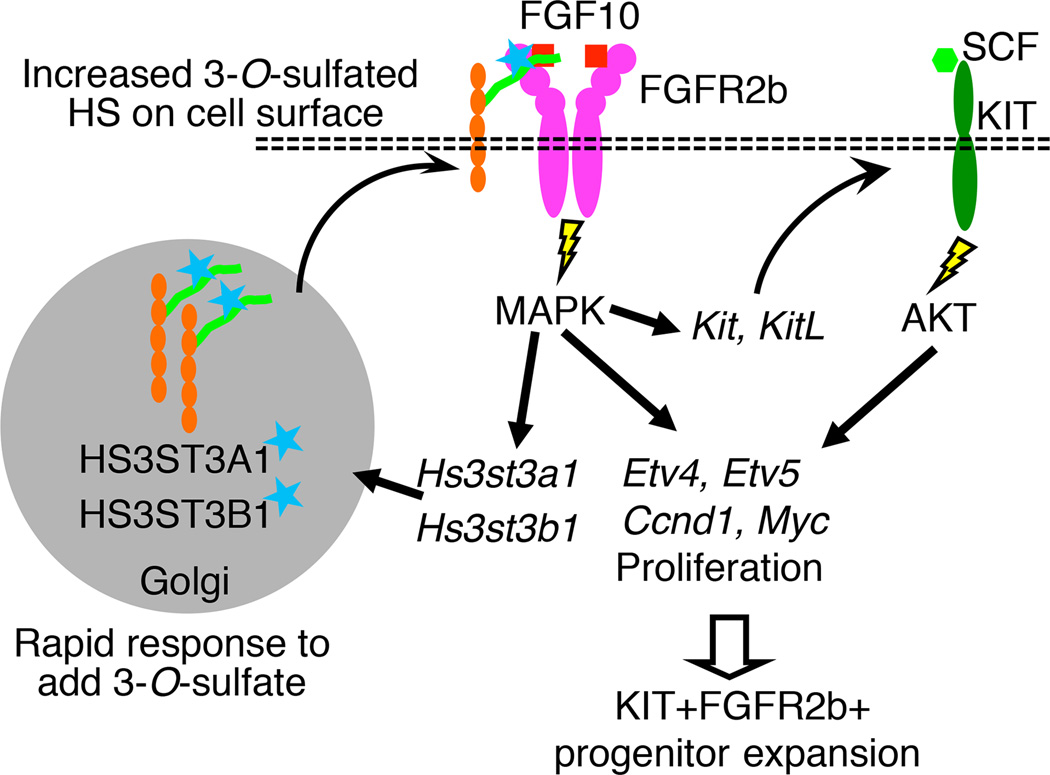
The exquisite control of growth factor function by heparan sulfate (HS) is dictated by tremendous structural heterogeneity of sulfated modifications. How specific HS structures control growth factor-dependent progenitor expansion during organogenesis is unknown. We isolated KIT+ progenitors from fetal salivary glands during a stage of rapid progenitor expansion and profiled HS biosynthetic enzyme expression. Enzymes generating a specific type of 3-O-sulfated-HS (3-O-HS) are enriched, and fibroblast growth factor 10 (FGF10)/FGF receptor 2b (FGFR2b) signaling directly regulates their expression. Bioengineered 3-O-HS binds FGFR2b and stabilizes FGF10/FGFR2b complexes in a receptor- and growth factor-specific manner. Rapid autocrine feedback increases 3-O-HS, KIT, and progenitor expansion. Knockdown of multiple Hs3st isoforms limits fetal progenitor expansion but is rescued with bioengineered 3-O-HS, which also increases adult progenitor expansion. Altering specific 3-O-sulfated epitopes provides a mechanism to rapidly respond to FGFR2b signaling and control progenitor expansion. 3-O-HS may expand KIT+ progenitors in vitro for regenerative therapy.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Belleudi F, Leone L, Nobili V, Raffa S, Francescangeli F, Maggio M, Morrone S, Marchese C, Torrisi MR. Keratinocyte growth factor receptor ligands target the receptor to different intracellular pathways. Traffic. 2007;8:1854–1872. - PubMed
-
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. - PubMed
-
- Bui C, Ouzzine M, Talhaoui I, Sharp S, Prydz K, Coughtrie MW, Fournel-Gigleux S. Epigenetics: methylation-associated repression of heparan sulfate 3-O-sulfotransferase gene expression contributes to the invasive phenotype of H-EMC-SS chondrosarcoma cells. FASEB J. 2010;24:436–450. - PubMed
-
- Cool SM, Nurcombe V. Heparan sulfate regulation of progenitor cell fate. J. Cell Biochem. 2006;99:1040–1051. - PubMed
-
- Davies JA. Do different branching epithelia use a conserved developmental mechanism? Bioessays. 2002;24:937–948. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
