

Defining a personal, allele-specific, and single-molecule long-read transcriptome
- PMID: 24961374
- PMCID: PMC4103364
- DOI: 10.1073/pnas.1400447111
Defining a personal, allele-specific, and single-molecule long-read transcriptome
Abstract
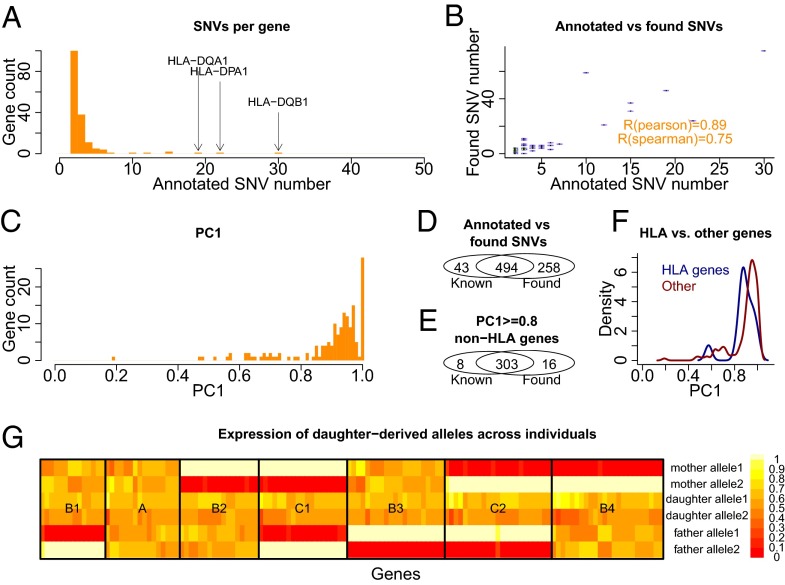
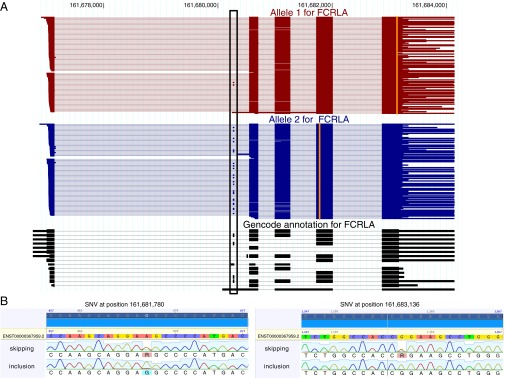
Personal transcriptomes in which all of an individual's genetic variants (e.g., single nucleotide variants) and transcript isoforms (transcription start sites, splice sites, and polyA sites) are defined and quantified for full-length transcripts are expected to be important for understanding individual biology and disease, but have not been described previously. To obtain such transcriptomes, we sequenced the lymphoblastoid transcriptomes of three family members (GM12878 and the parents GM12891 and GM12892) by using a Pacific Biosciences long-read approach complemented with Illumina 101-bp sequencing and made the following observations. First, we found that reads representing all splice sites of a transcript are evident for most sufficiently expressed genes ≤3 kb and often for genes longer than that. Second, we added and quantified previously unidentified splicing isoforms to an existing annotation, thus creating the first personalized annotation to our knowledge. Third, we determined SNVs in a de novo manner and connected them to RNA haplotypes, including HLA haplotypes, thereby assigning single full-length RNA molecules to their transcribed allele, and demonstrated Mendelian inheritance of RNA molecules. Fourth, we show how RNA molecules can be linked to personal variants on a one-by-one basis, which allows us to assess differential allelic expression (DAE) and differential allelic isoforms (DAI) from the phased full-length isoform reads. The DAI method is largely independent of the distance between exon and SNV--in contrast to fragmentation-based methods. Overall, in addition to improving eukaryotic transcriptome annotation, these results describe, to our knowledge, the first large-scale and full-length personal transcriptome.
Keywords: allele-specific expression; alternative splicing; isoform sequencing; personalized medicine; platform comparison.
Conflict of interest statement
Conflict of interest statement: M.P.S. is on the scientific advisory board of Personalis and GenapSys.
Figures
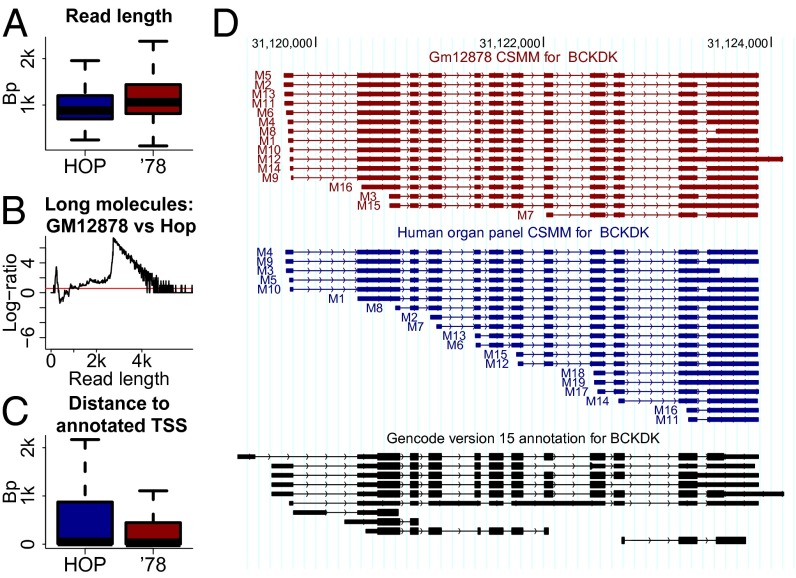
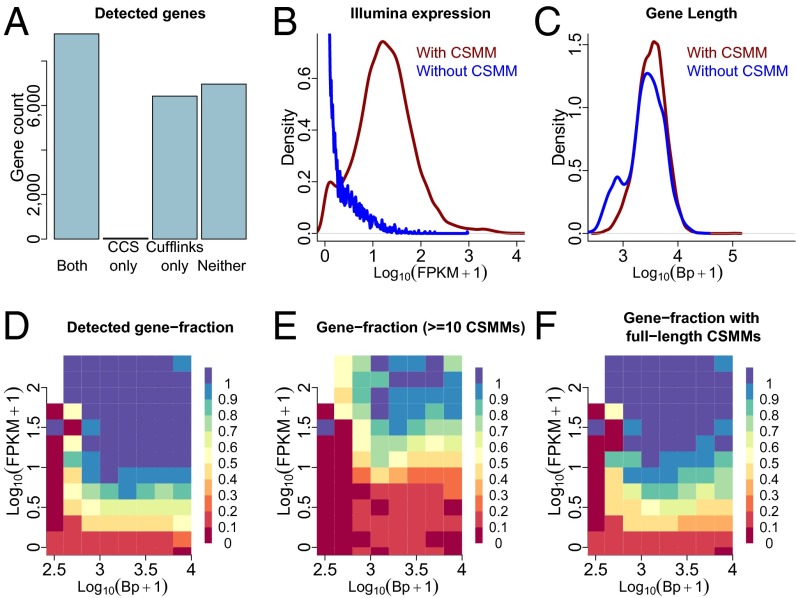
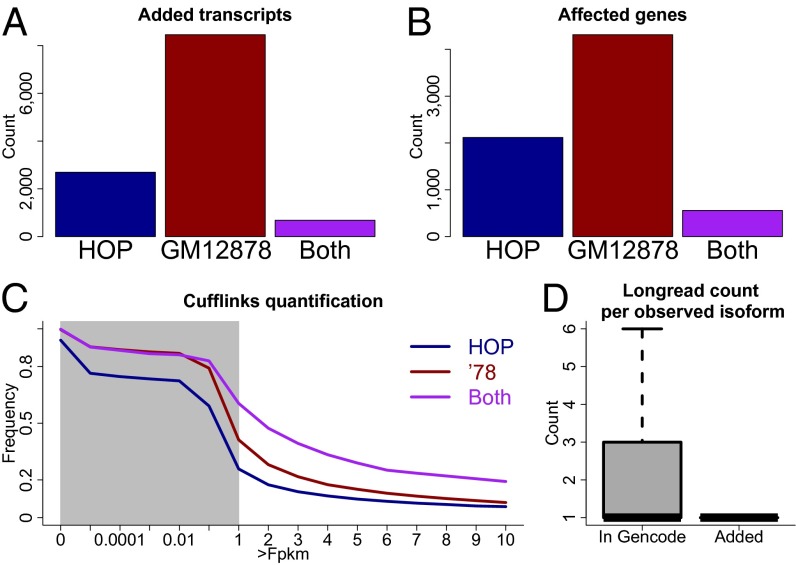
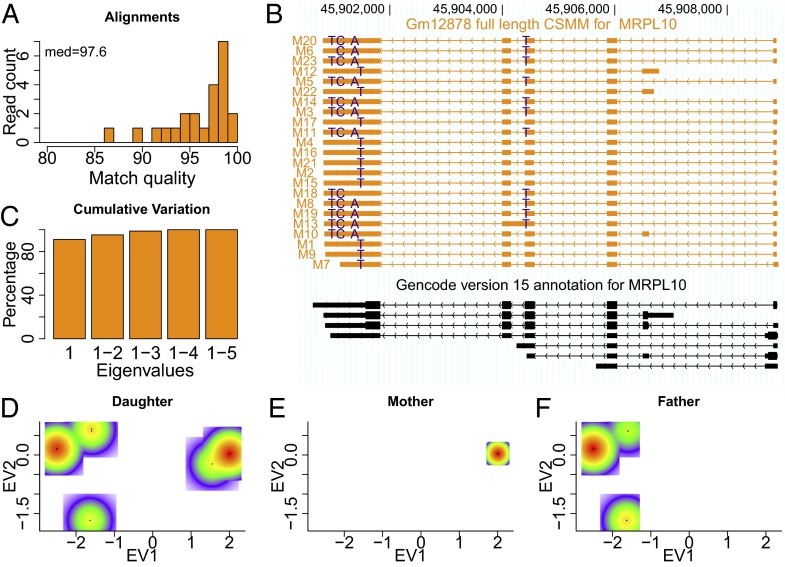
References
-
- Sultan M, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–960. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. - PubMed
-
- Wilhelm BT, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–1243. - PubMed
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
