

Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion
- PMID: 24961702
- PMCID: PMC4066239
- DOI: 10.3390/brainsci4010091
Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion
Abstract
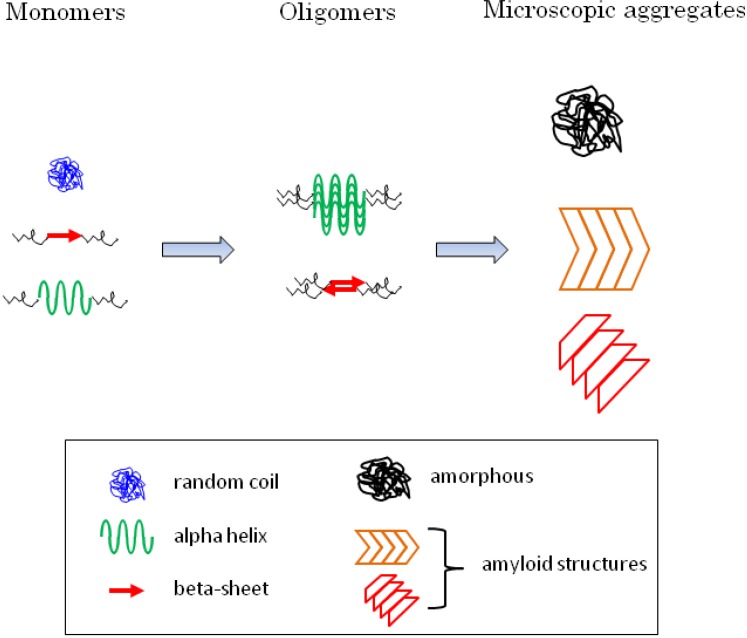
Huntington disease and other diseases of polyglutamine expansion are each caused by a different protein bearing an excessively long polyglutamine sequence and are associated with neuronal death. Although these diseases affect largely different brain regions, they all share a number of characteristics, and, therefore, are likely to possess a common mechanism. In all of the diseases, the causative protein is proteolyzed, becomes abnormally folded and accumulates in oligomers and larger aggregates. The aggregated and possibly the monomeric expanded polyglutamine are likely to play a critical role in the pathogenesis and there is increasing evidence that the secondary structure of the protein influences its toxicity. We describe here, with special attention to huntingtin, the mechanisms of polyglutamine aggregation and the modulation of aggregation by the sequences flanking the polyglutamine. We give a comprehensive picture of the characteristics of monomeric and aggregated polyglutamine, including morphology, composition, seeding ability, secondary structure, and toxicity. The structural heterogeneity of aggregated polyglutamine may explain why polyglutamine-containing aggregates could paradoxically be either toxic or neuroprotective.
Figures
References
-
- Sanpei K., Takano H., Igarashi S., Sato T., Oyake M., Sasaki H., Wakisaka A., Tashiro K., Ishida Y., Ikeuchi T., et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, direct. Nat. Genet. 1996;14:277–284. doi: 10.1038/ng1196-277. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
