

Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth
- PMID: 24963049
- PMCID: PMC4155010
- DOI: 10.1158/1078-0432.CCR-14-0072
Reversible LSD1 inhibition interferes with global EWS/ETS transcriptional activity and impedes Ewing sarcoma tumor growth
Abstract
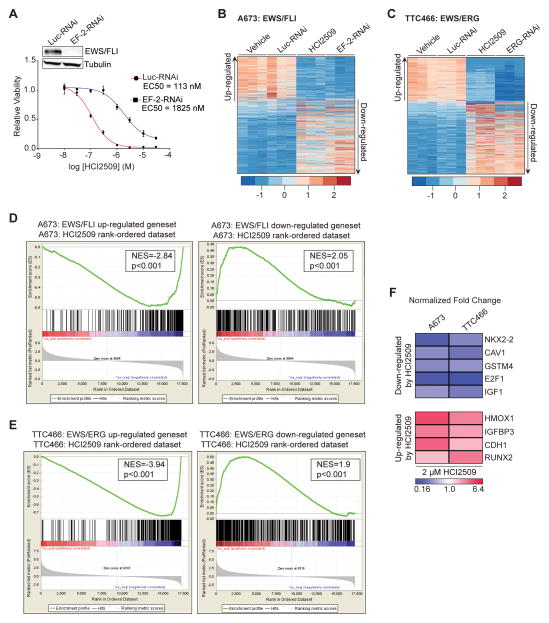
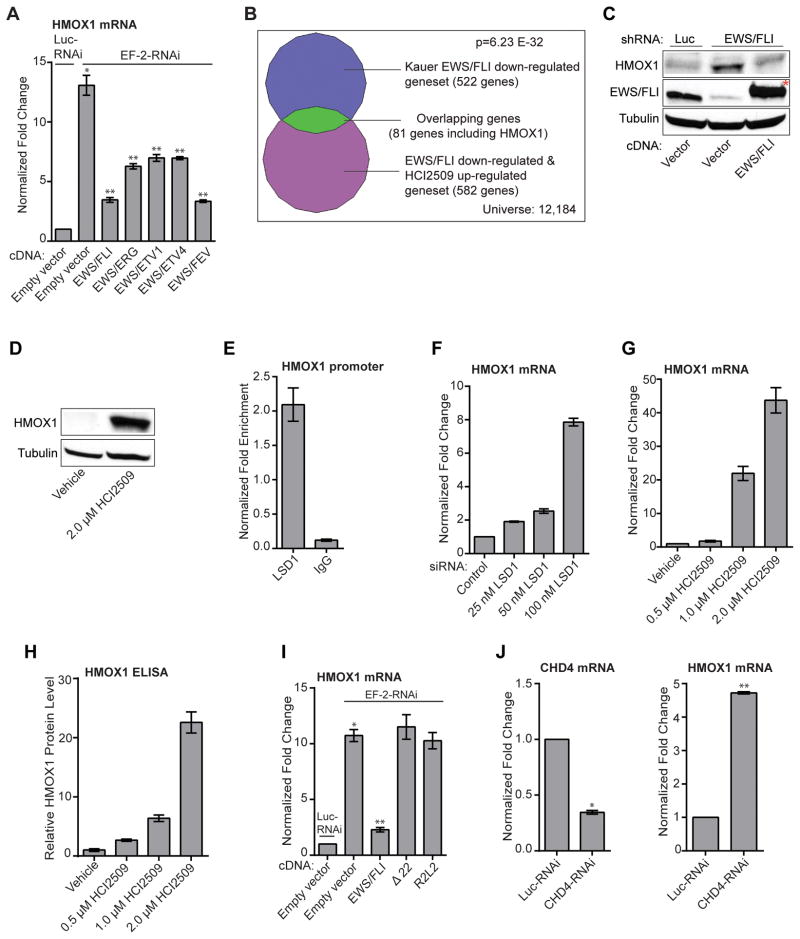
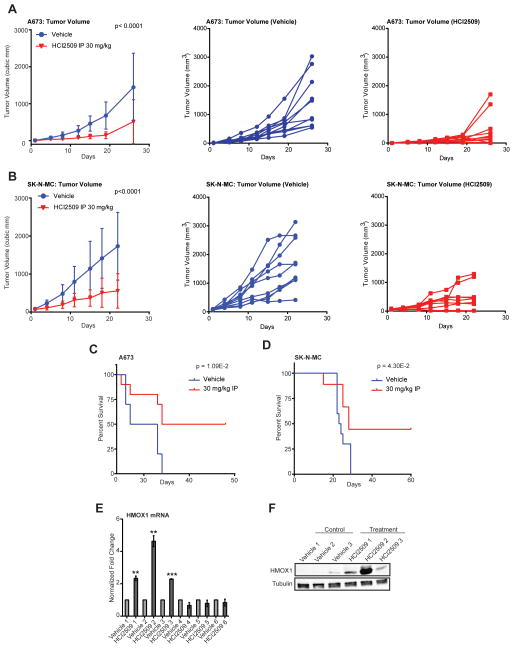
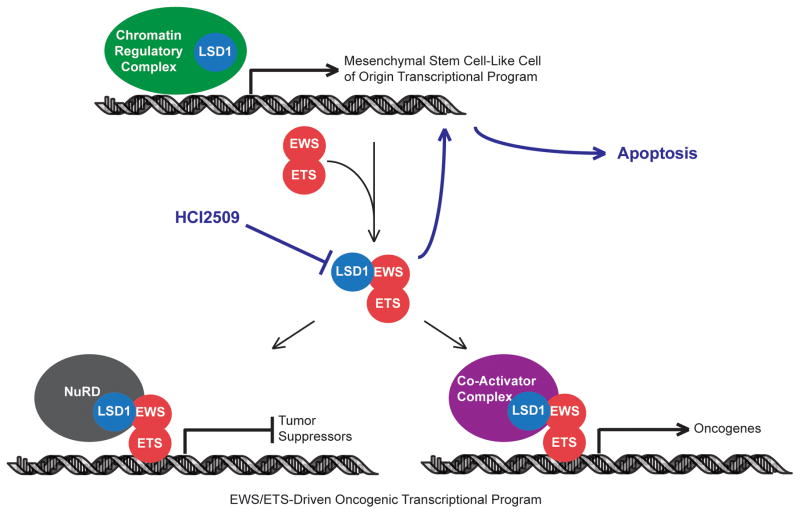
Purpose: Ewing sarcoma is a pediatric bone tumor that absolutely relies on the transcriptional activity of the EWS/ETS family of fusion oncoproteins. While the most common fusion, EWS/FLI, utilizes lysine-specific demethylase 1 (LSD1) to repress critical tumor suppressors, small-molecule blockade of LSD1 has not yet been thoroughly explored as a therapeutic approach for Ewing sarcoma. We therefore evaluated the translational potential of potent and specific LSD1 inhibition with HCI2509 on the transcriptional program of both EWS/FLI and EWS/ERG as well as the downstream oncogenic phenotypes driven by EWS/ETS fusions in both in vitro and in vivo models of Ewing sarcoma.
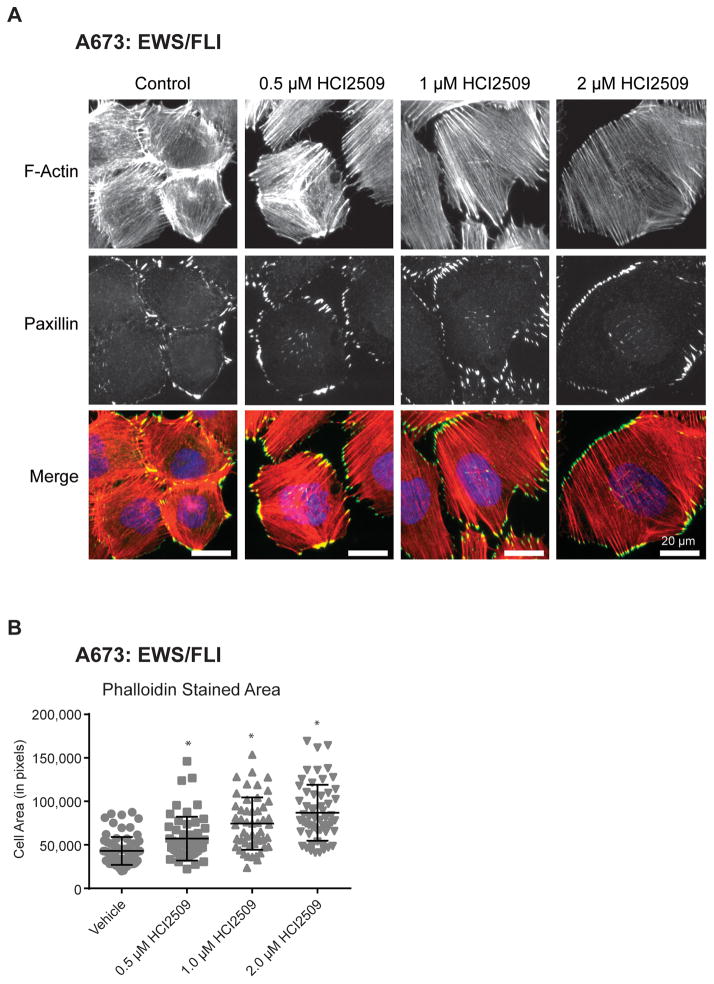
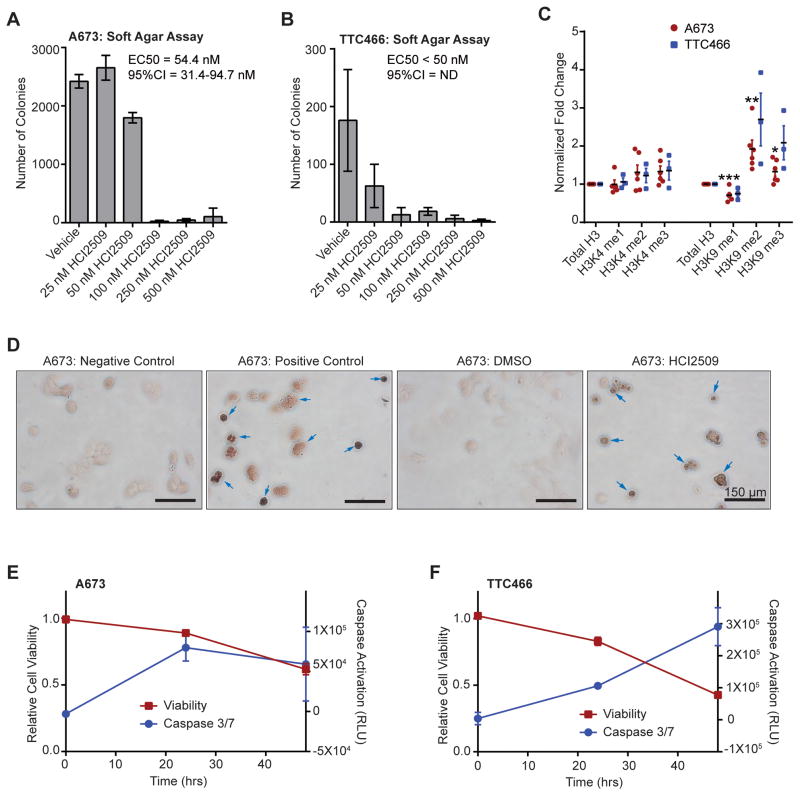
Experimental design: RNA-seq was used to compare the transcriptional profiles of EWS/FLI, EWS/ERG, and treatment with HCI2509 in both EWS/FLI- and EWS/ERG-containing cell lines. We then evaluated morphologic phenotypes of treated cells with immunofluorescence. The induction of apoptosis was evaluated using caspase-3/7 activation and TUNEL staining. Colony forming assays were used to test oncogenic transformation and xenograft studies with patient-derived cell lines were used to evaluate the effects of HCI2509 on tumorigenesis.
Results: HCI2509 caused a dramatic reversal of both the up- and downregulated transcriptional profiles of EWS/FLI and EWS/ERG accompanied by the induction of apoptosis and disruption of morphologic and oncogenic phenotypes modulated by EWS/FLI. Importantly, HCI2509 displayed single-agent efficacy in multiple xenograft models.
Conclusions: These data support epigenetic modulation with HCI2509 as a therapeutic strategy for Ewing sarcoma, and highlight a critical dual role for LSD1 in the oncogenic transcriptional activity of EWS/ETS proteins.
©2014 American Association for Cancer Research.
Conflict of interest statement
Figures
References
-
- Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. - PubMed
-
- Lachner M, O’Sullivan RJ, Jenuwein T. An epigenetic road map for histone lysine methylation. J Cell Sci. 2003;116:2117–24. - PubMed
-
- Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. - PubMed
-
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
