

WONOEP appraisal: new genetic approaches to study epilepsy
- PMID: 24965021
- PMCID: PMC4126888
- DOI: 10.1111/epi.12692
WONOEP appraisal: new genetic approaches to study epilepsy
Abstract
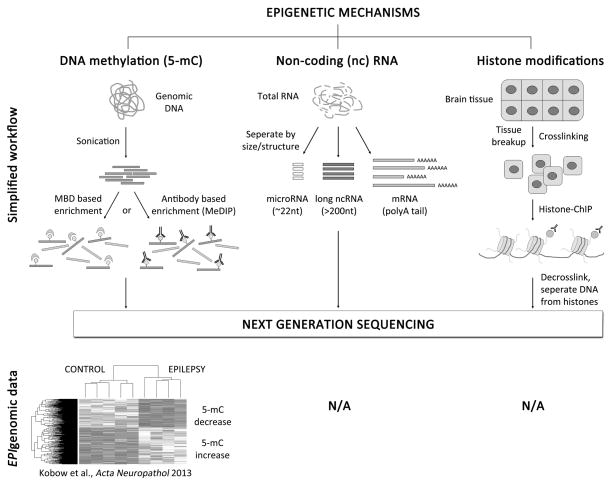
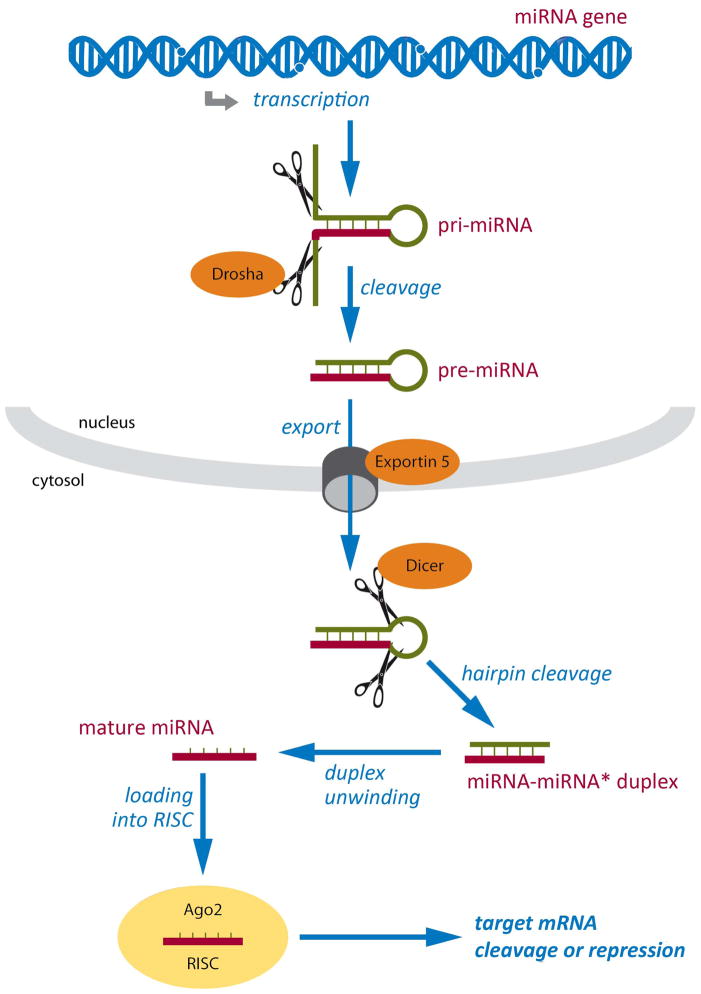
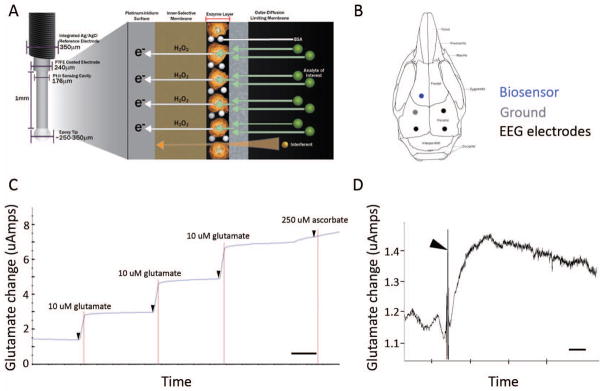
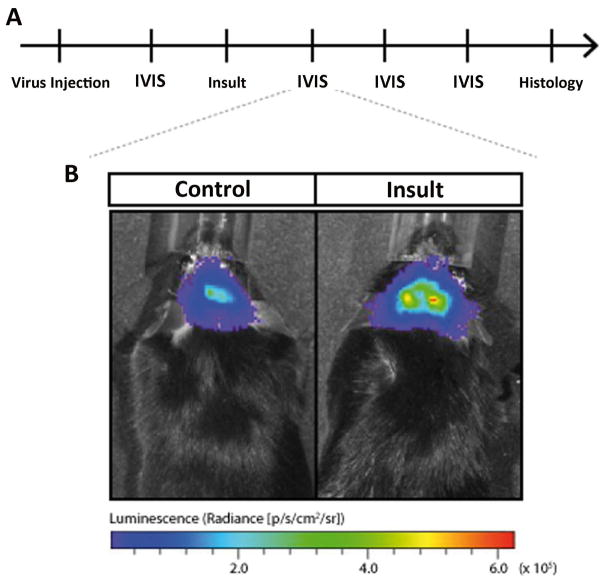
New genetic investigation techniques, including next-generation sequencing, epigenetic profiling, cell lineage mapping, targeted genetic manipulation of specific neuronal cell types, stem cell reprogramming, and optogenetic manipulations within epileptic networks are progressively unraveling the mysteries of epileptogenesis and ictogenesis. These techniques have opened new avenues to discover the molecular basis of epileptogenesis and to study the physiologic effects of mutations in epilepsy-associated genes on a multilayer level, from cells to circuits. This manuscript reviews recently published applications of these new genetic technologies in the study of epilepsy, as well as work presented by the authors at the genetic session of the XII Workshop on the Neurobiology of Epilepsy (WONOEP 2013) in Quebec, Canada. Next-generation sequencing is providing investigators with an unbiased means to assess the molecular causes of sporadic forms of epilepsy and has revealed the complexity and genetic heterogeneity of sporadic epilepsy disorders. To assess the functional impact of mutations in these newly identified genes on specific neuronal cell types during brain development, new modeling strategies in animals, including conditional genetics in mice and in utero knock-down approaches, are enabling functional validation with exquisite cell-type and temporal specificity. In addition, optogenetics, using cell-type-specific Cre recombinase driver lines, is enabling investigators to dissect networks involved in epilepsy. In addition, genetically encoded cell-type labeling is providing new means to assess the role of the nonneuronal components of epileptic networks such as glial cells. Furthermore, beyond its role in revealing coding variants involved in epileptogenesis, next-generation sequencing can be used to assess the epigenetic modifications that lead to sustained network hyperexcitability in epilepsy, including methylation changes in gene promoters and noncoding ribonucleic acid (RNA) involved in modifying gene expression following seizures. In addition, genetically based bioluminescent reporters are providing new opportunities to assess neuronal activity and neurotransmitter levels both in vitro and in vivo in the context of epilepsy. Finally, genetically rederived neurons generated from patient induced pluripotent stem cells and genetically modified zebrafish have become high-throughput means to investigate disease mechanisms and potential new therapies. Genetics has changed the field of epilepsy research considerably, and is paving the way for better diagnosis and therapies for patients with epilepsy.
Keywords: Calcium channels; Cytoskeleton; EEG monitoring; Interneurons; Systems biology; “-Omics”.
Wiley Periodicals, Inc. © 2014 International League Against Epilepsy.
Figures
References
-
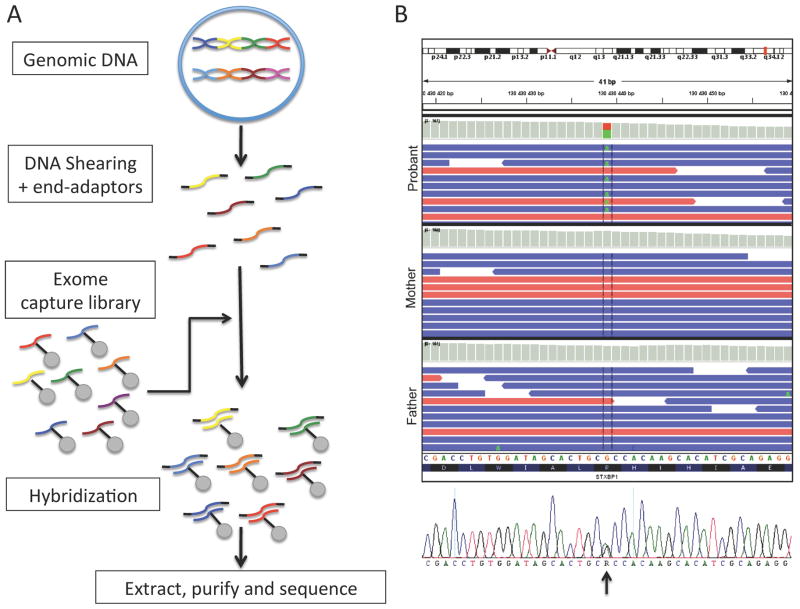
- Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
