

Structure of a modular polyketide synthase
- PMID: 24965652
- PMCID: PMC4278352
- DOI: 10.1038/nature13423
Structure of a modular polyketide synthase
Abstract
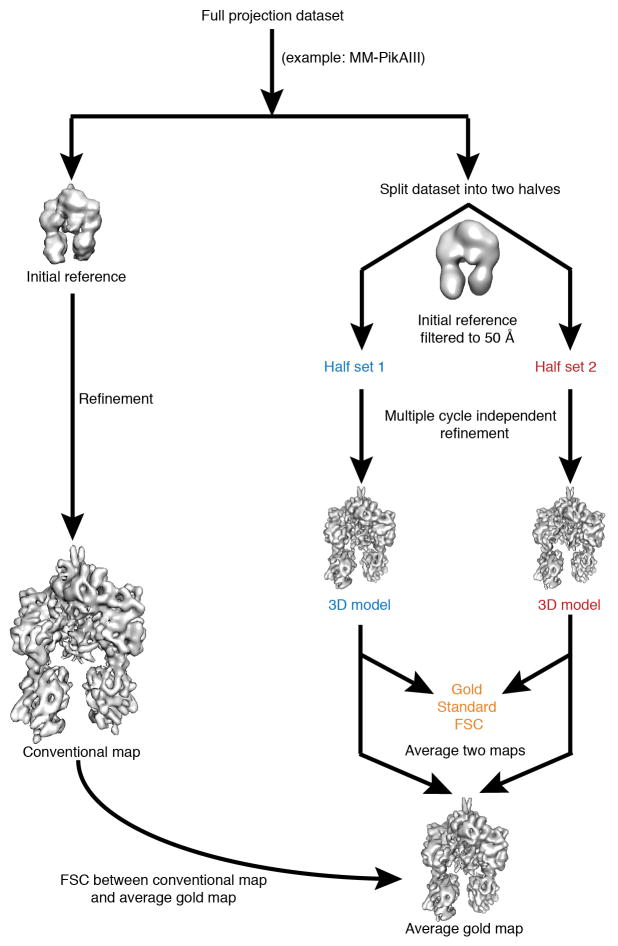
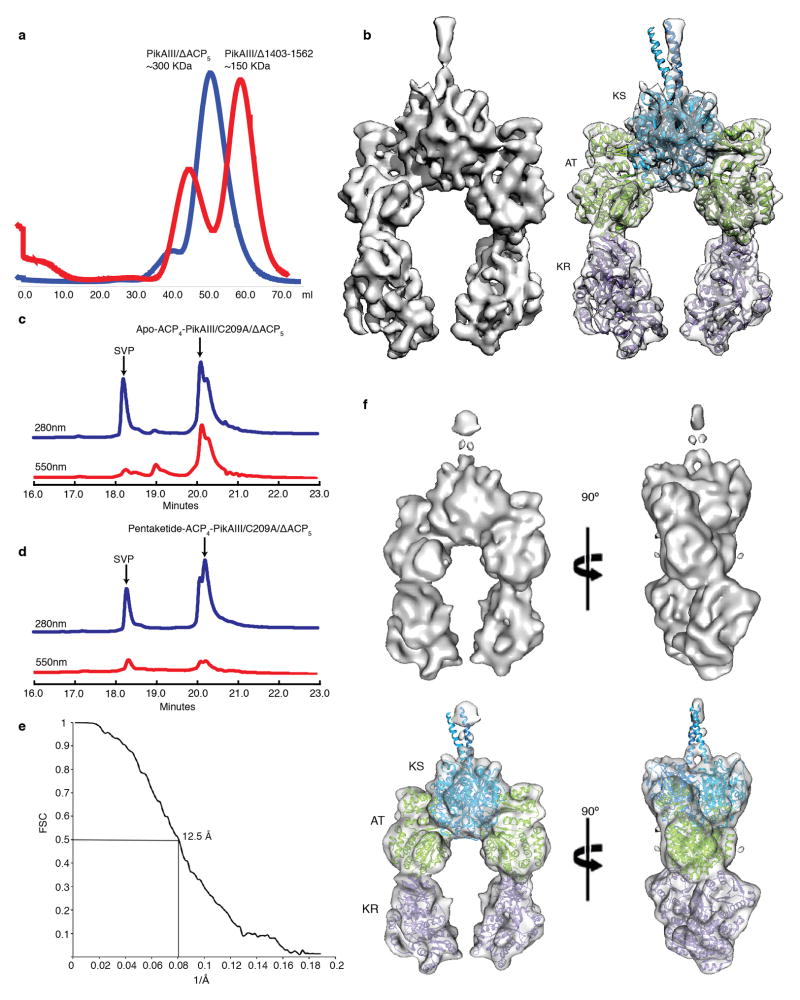
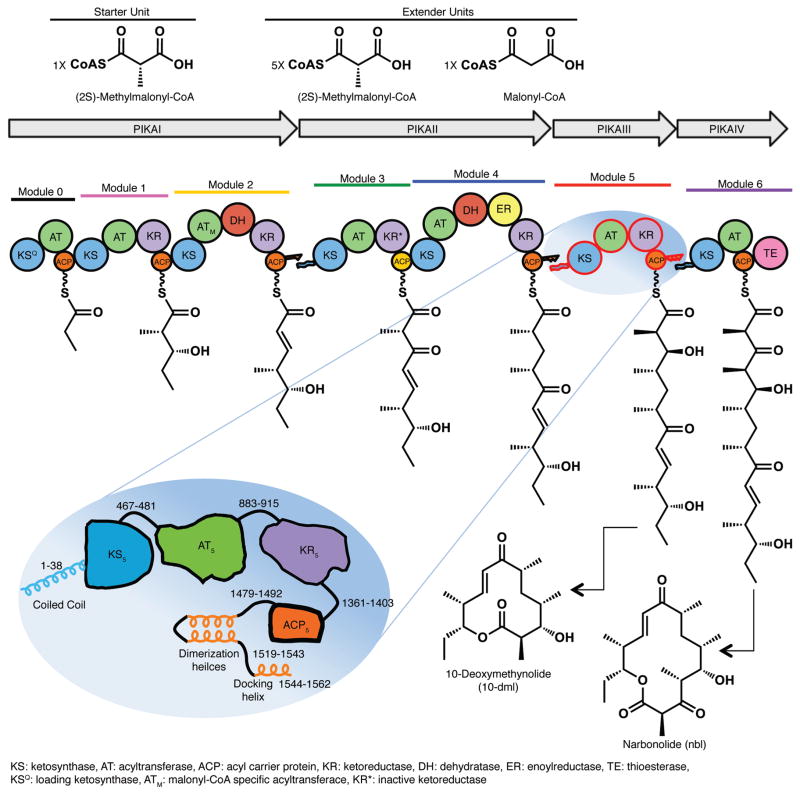
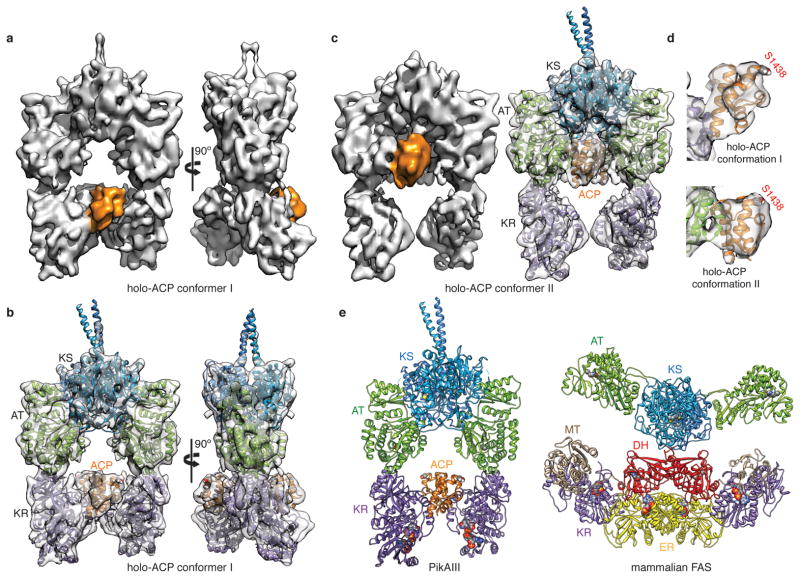
Polyketide natural products constitute a broad class of compounds with diverse structural features and biological activities. Their biosynthetic machinery, represented by type I polyketide synthases (PKSs), has an architecture in which successive modules catalyse two-carbon linear extensions and keto-group processing reactions on intermediates covalently tethered to carrier domains. Here we used electron cryo-microscopy to determine sub-nanometre-resolution three-dimensional reconstructions of a full-length PKS module from the bacterium Streptomyces venezuelae that revealed an unexpectedly different architecture compared to the homologous dimeric mammalian fatty acid synthase. A single reaction chamber provides access to all catalytic sites for the intramodule carrier domain. In contrast, the carrier from the preceding module uses a separate entrance outside the reaction chamber to deliver the upstream polyketide intermediate for subsequent extension and modification. This study reveals for the first time, to our knowledge, the structural basis for both intramodule and intermodule substrate transfer in polyketide synthases, and establishes a new model for molecular dissection of these multifunctional enzyme systems.
Figures
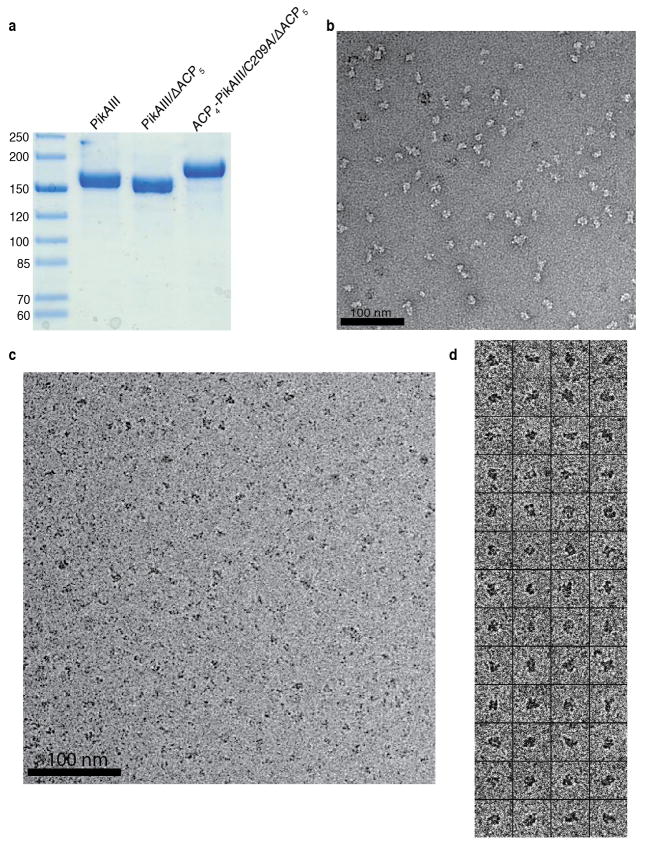
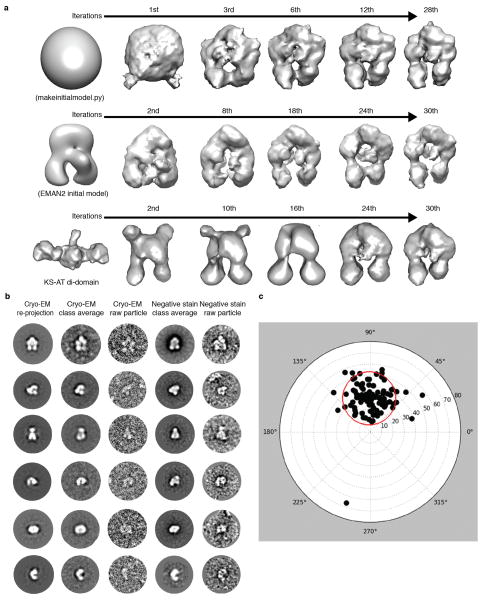
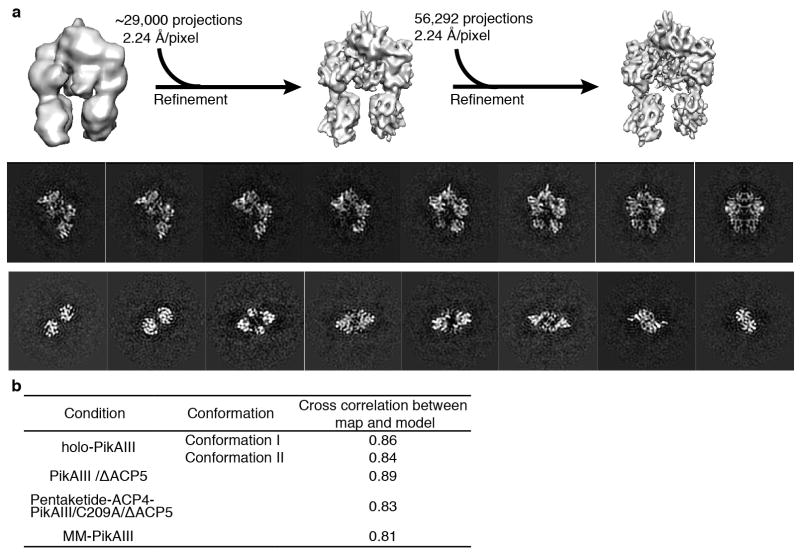
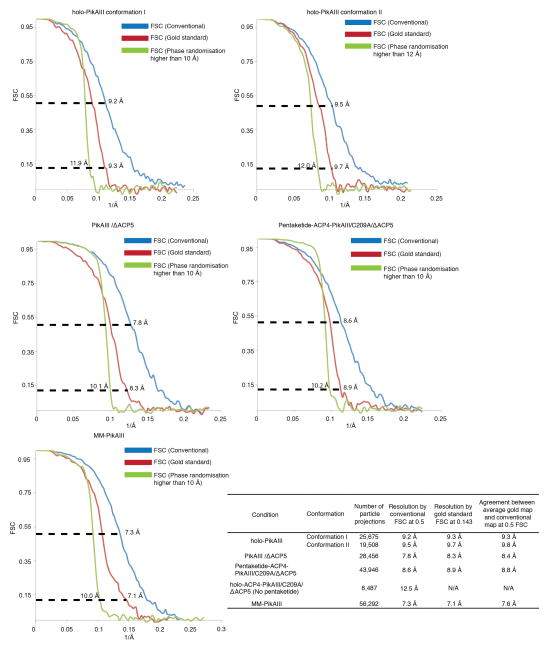
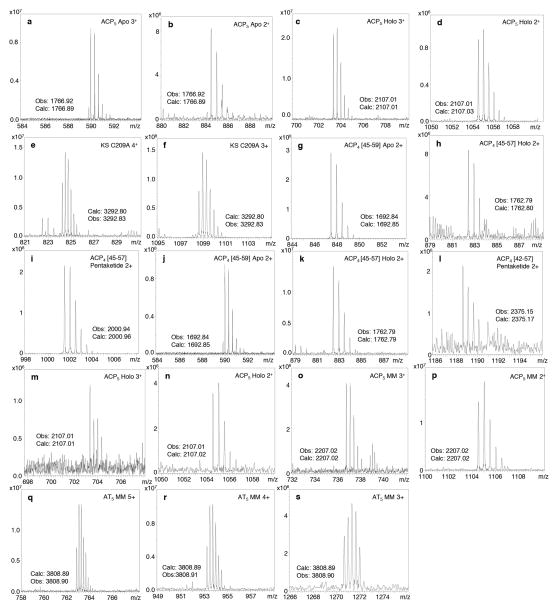
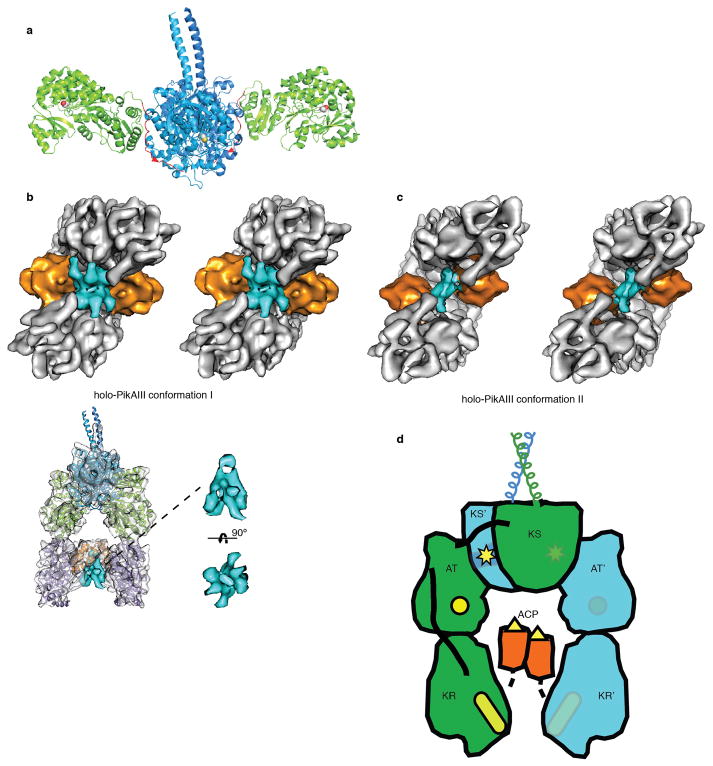
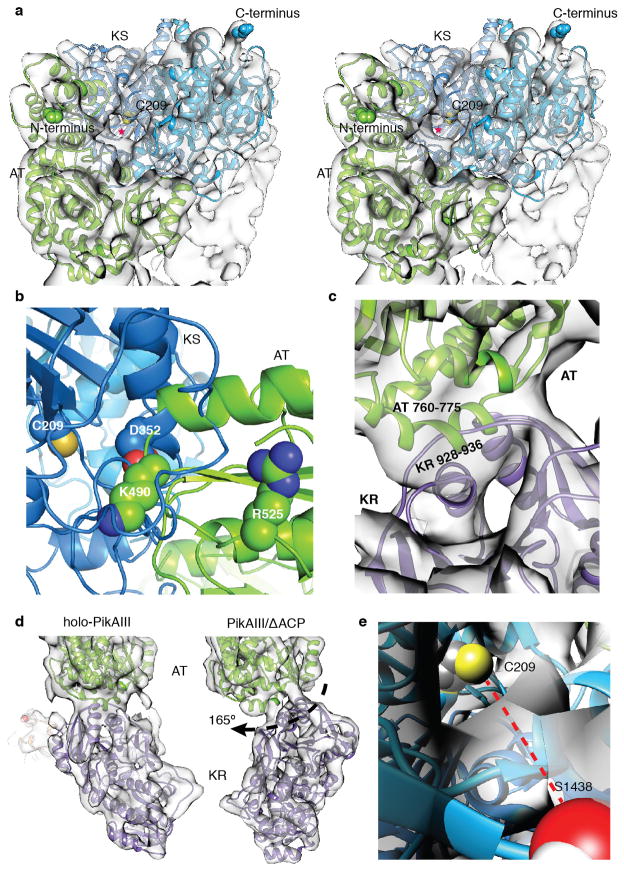
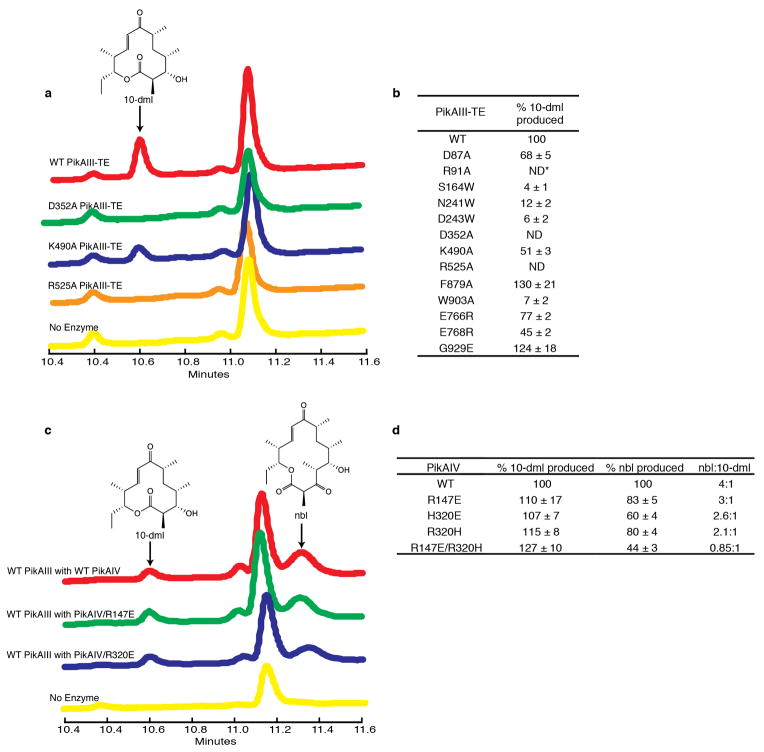
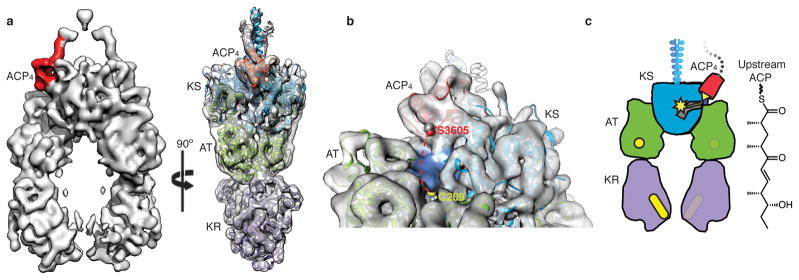
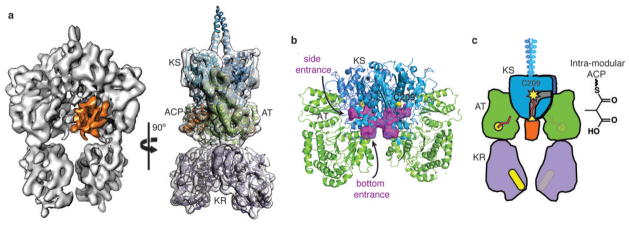
Comment in
-
Structural biology: enzyme assembly line pictured.Nature. 2014 Jun 26;510(7506):482-3. doi: 10.1038/nature13505. Epub 2014 Jun 18. Nature. 2014. PMID: 24965648 No abstract available.
References
-
- Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. - PubMed
-
- Broadhurst RW, Nietlispach D, Wheatcroft MP, Leadlay PF, Weissman KJ. The structure of docking domains in modular polyketide synthases. Chem Biol. 2003;10:723–731. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
