

Emerging role of the β-catenin-PPARγ axis in the pathogenesis of colorectal cancer
- PMID: 24966585
- PMCID: PMC4064060
- DOI: 10.3748/wjg.v20.i23.7137
Emerging role of the β-catenin-PPARγ axis in the pathogenesis of colorectal cancer
Abstract
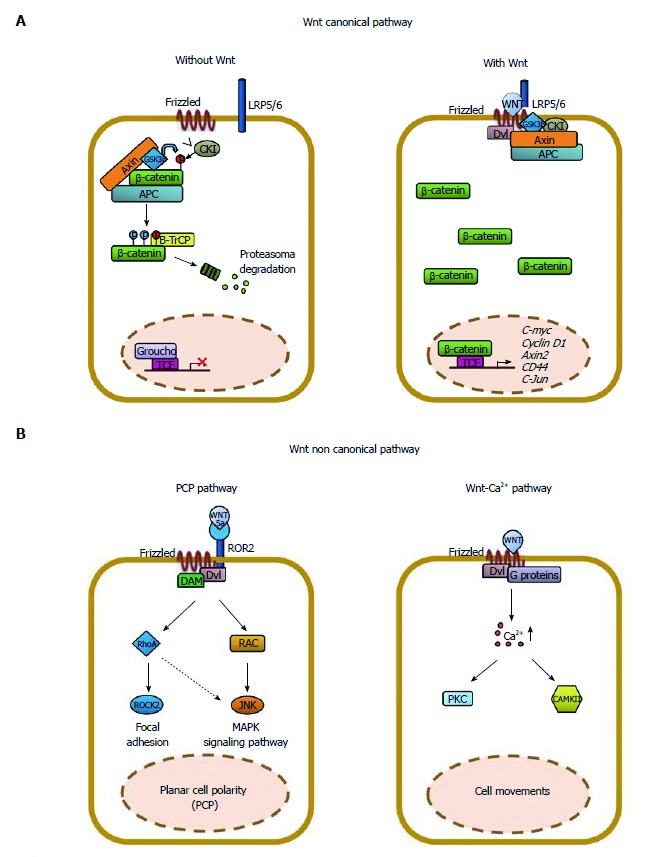
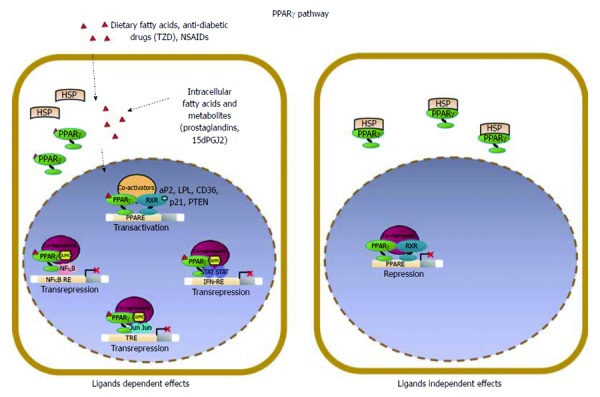
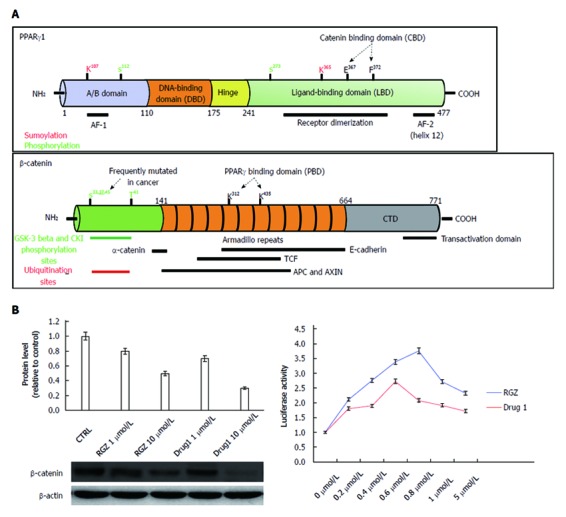
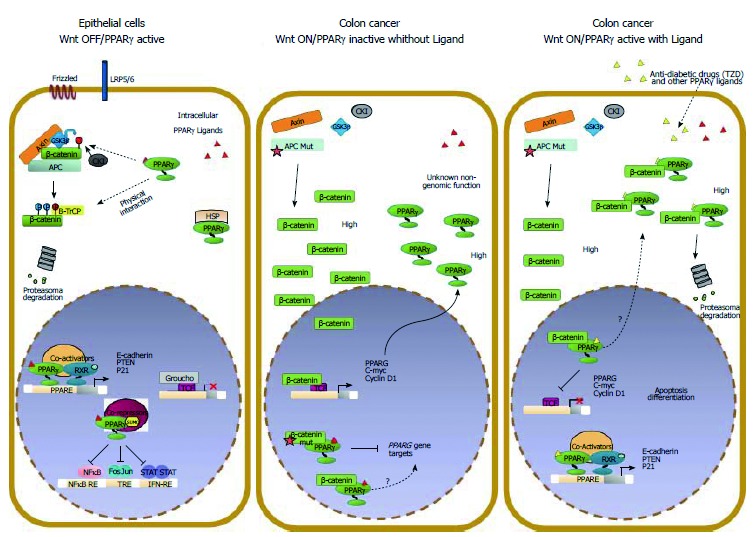
Multiple lines of evidence indicate that Wnt/β-catenin signaling plays a fundamental role in colorectal cancer (CRC) initiation and progression. Recent genome-wide data have confirmed that in CRC this pathway is one of the most frequently modified by genetic or epigenetic alterations affecting almost 90% of Wnt/β-catenin gene members. A major challenge is thus learning how the corrupted coordination of this pathway is tied to other signalings to enhance cell growth. Peroxisome proliferator activated receptor γ (PPARγ) is emerging as a growth-limiting and differentiation-promoting factor. In tumorigenesis it exerts a tumor suppressor role and is potentially linked with the Wnt/β-catenin pathway. Based on these results, the identification of new selective PPARγ modulators with inhibitory effects on the Wnt/β-catenin pathway is becoming an interesting perspective. Should, in fact, these molecules display such properties, new research avenues would be opened aimed at developing new molecular targeted drugs. Herein, we review the basic principles and present new hypotheses underlying the crosstalk between Wnt/β-catenin and PPARγ signaling. Furthermore, we discuss the advances in our understanding as to how their altered regulation can culminate in colon cancer and the efforts aimed at designing novel PPARγ agonists endowed with Wnt/β-catenin inhibitory effects to be used as therapeutic and/or preventive agents.
Keywords: Colorectal cancer; Genomic instability; Peroxisome proliferator activated receptor γ ligands; Peroxisome proliferator-activated receptor γ; Wnt/β-catenin pathway.
Figures
References
-
- Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. - PubMed
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. - PubMed
-
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
