

Aberrant autolysosomal regulation is linked to the induction of embryonic senescence: differential roles of Beclin 1 and p53 in vertebrate Spns1 deficiency
- PMID: 24967584
- PMCID: PMC4072523
- DOI: 10.1371/journal.pgen.1004409
Aberrant autolysosomal regulation is linked to the induction of embryonic senescence: differential roles of Beclin 1 and p53 in vertebrate Spns1 deficiency
Abstract
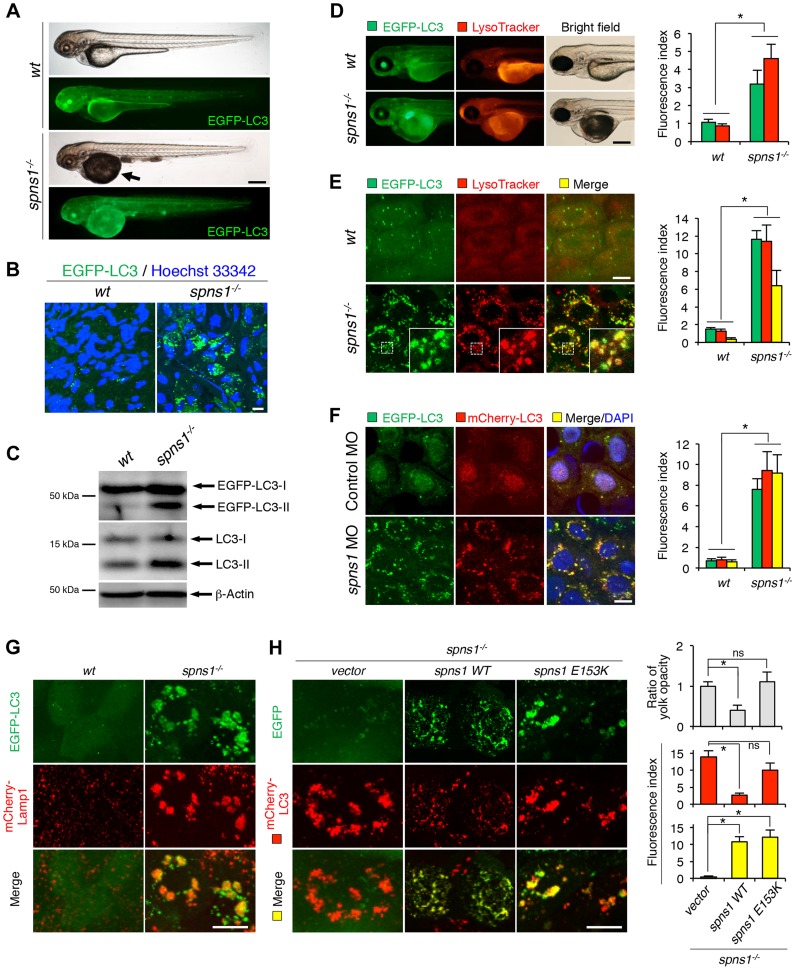
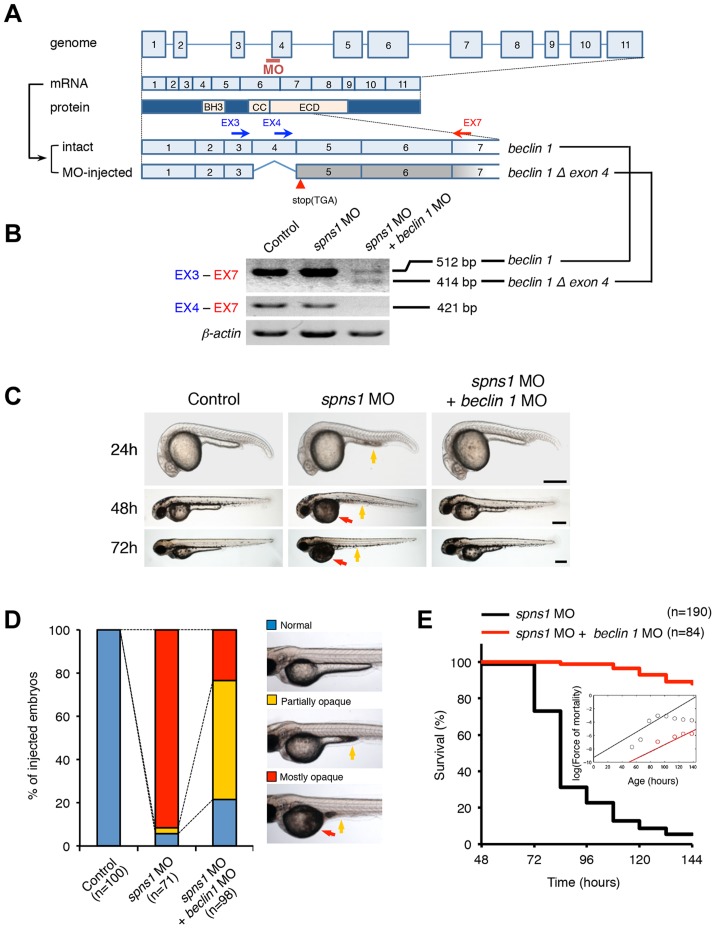
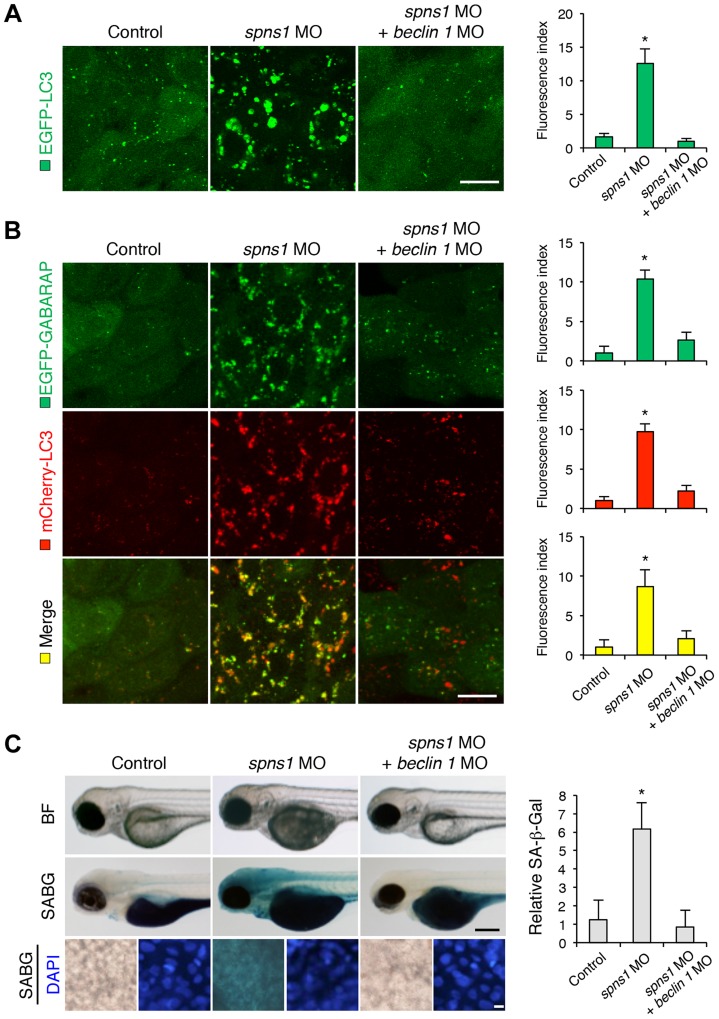
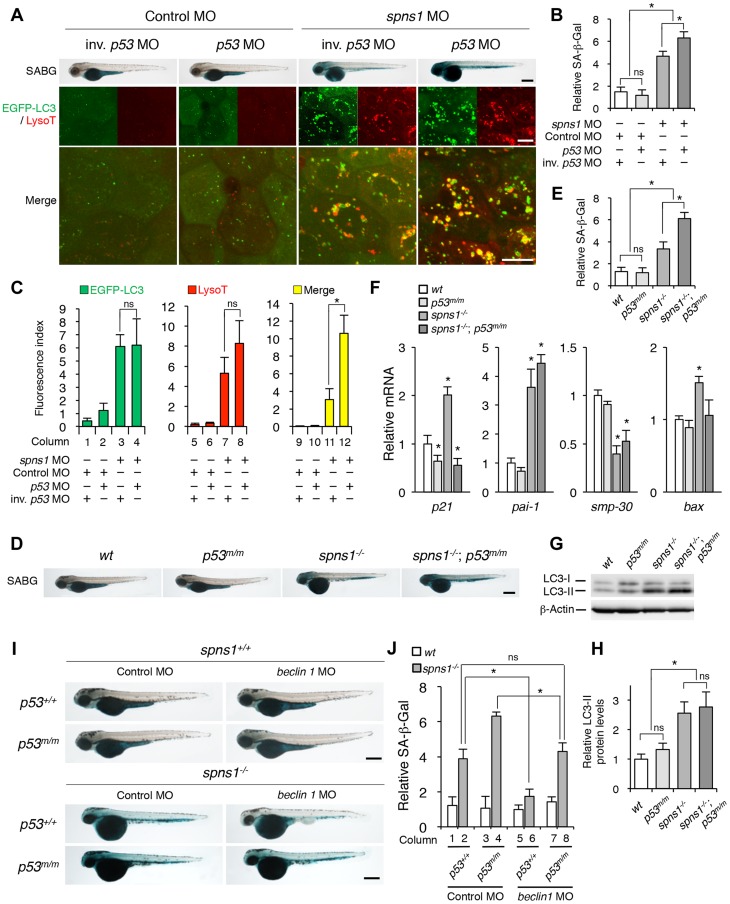
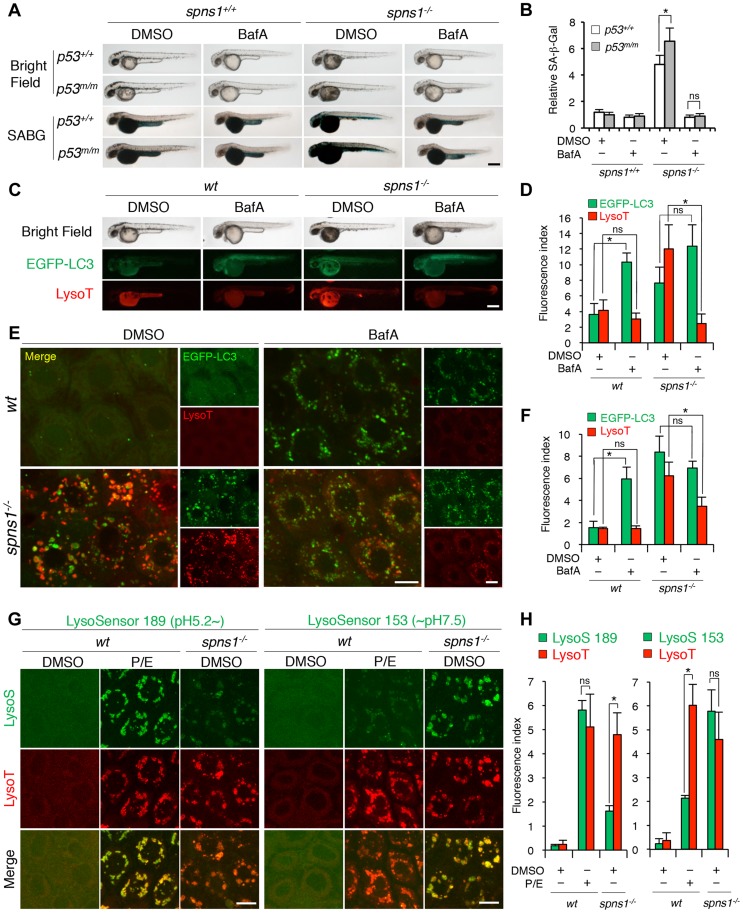
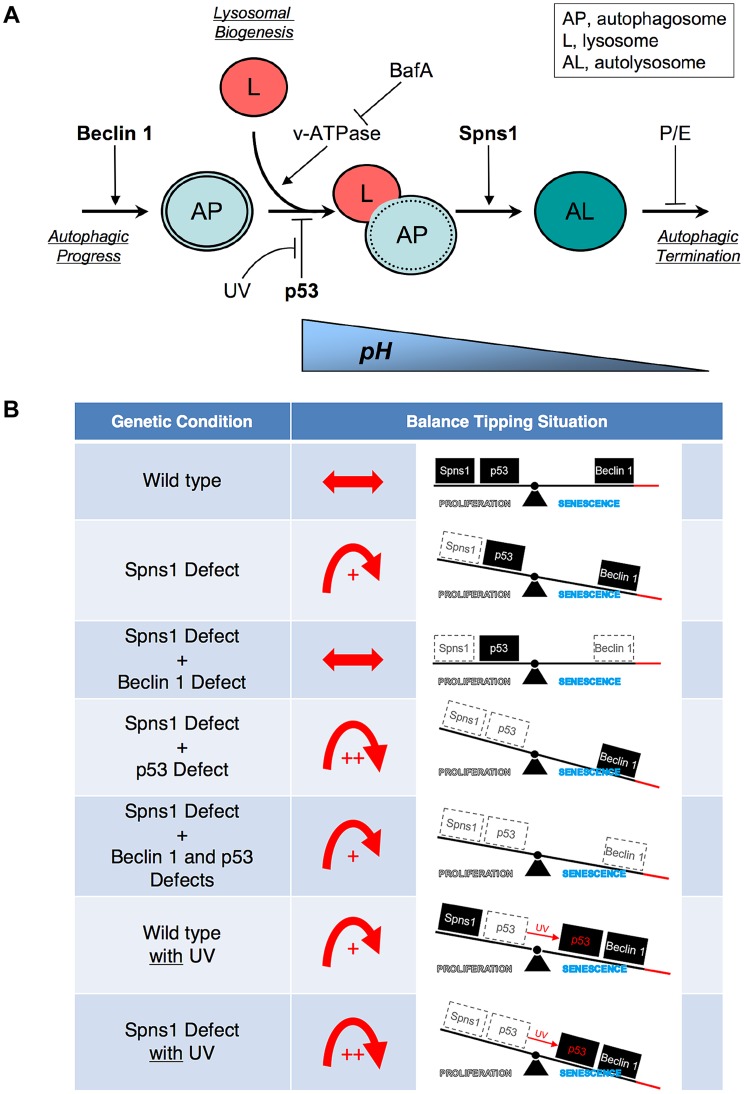
Spinster (Spin) in Drosophila or Spinster homolog 1 (Spns1) in vertebrates is a putative lysosomal H+-carbohydrate transporter, which functions at a late stage of autophagy. The Spin/Spns1 defect induces aberrant autolysosome formation that leads to embryonic senescence and accelerated aging symptoms, but little is known about the mechanisms leading to the pathogenesis in vivo. Beclin 1 and p53 are two pivotal tumor suppressors that are critically involved in the autophagic process and its regulation. Using zebrafish as a genetic model, we show that Beclin 1 suppression ameliorates Spns1 loss-mediated senescence as well as autophagic impairment, whereas unexpectedly p53 deficit exacerbates both of these characteristics. We demonstrate that 'basal p53' activity plays a certain protective role(s) against the Spns1 defect-induced senescence via suppressing autophagy, lysosomal biogenesis, and subsequent autolysosomal formation and maturation, and that p53 loss can counteract the effect of Beclin 1 suppression to rescue the Spns1 defect. By contrast, in response to DNA damage, 'activated p53' showed an apparent enhancement of the Spns1-deficient phenotype, by inducing both autophagy and apoptosis. Moreover, we found that a chemical and genetic blockage of lysosomal acidification and biogenesis mediated by the vacuolar-type H+-ATPase, as well as of subsequent autophagosome-lysosome fusion, prevents the appearance of the hallmarks caused by the Spns1 deficiency, irrespective of the basal p53 state. Thus, these results provide evidence that Spns1 operates during autophagy and senescence differentially with Beclin 1 and p53.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Carbofuran accelerates the cellular senescence and declines the life span of spns1 mutant zebrafish.J Cell Mol Med. 2021 Jan;25(2):1048-1059. doi: 10.1111/jcmm.16171. Epub 2020 Dec 4. J Cell Mol Med. 2021. PMID: 33277797 Free PMC article.
-
Autolysosome biogenesis and developmental senescence are regulated by both Spns1 and v-ATPase.Autophagy. 2017 Feb;13(2):386-403. doi: 10.1080/15548627.2016.1256934. Epub 2016 Nov 22. Autophagy. 2017. PMID: 27875093 Free PMC article.
-
Carbofuran affects cellular autophagy and developmental senescence through the impairment of Nrf2 signalling.J Cell Mol Med. 2022 Jan;26(1):35-47. doi: 10.1111/jcmm.16774. Epub 2021 Jul 9. J Cell Mol Med. 2022. PMID: 34240810 Free PMC article.
-
Stories of spinster with various faces: from courtship rejection to tumor metastasis rejection.J Neurogenet. 2019 Mar-Jun;33(2):90-95. doi: 10.1080/01677063.2019.1586897. Epub 2019 Apr 2. J Neurogenet. 2019. PMID: 30939968 Review.
-
Impaired autophagy and APP processing in Alzheimer's disease: The potential role of Beclin 1 interactome.Prog Neurobiol. 2013 Jul-Aug;106-107:33-54. doi: 10.1016/j.pneurobio.2013.06.002. Epub 2013 Jul 1. Prog Neurobiol. 2013. PMID: 23827971 Review.
Cited by
-
Ultraspecific live imaging of the dynamics of zebrafish neutrophil granules by a histopermeable fluorogenic benzochalcone probe.Chem Sci. 2019 Feb 14;10(12):3654-3670. doi: 10.1039/c8sc05593a. eCollection 2019 Mar 28. Chem Sci. 2019. PMID: 30996961 Free PMC article.
-
Molecular characterization of Beclin 1 in rare minnow (Gobiocypris rarus) and its expression after waterborne cadmium exposure.Fish Physiol Biochem. 2016 Feb;42(1):111-23. doi: 10.1007/s10695-015-0122-1. Epub 2015 Sep 7. Fish Physiol Biochem. 2016. PMID: 26347097
-
Carbofuran accelerates the cellular senescence and declines the life span of spns1 mutant zebrafish.J Cell Mol Med. 2021 Jan;25(2):1048-1059. doi: 10.1111/jcmm.16171. Epub 2020 Dec 4. J Cell Mol Med. 2021. PMID: 33277797 Free PMC article.
-
Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition).Autophagy. 2016;12(1):1-222. doi: 10.1080/15548627.2015.1100356. Autophagy. 2016. PMID: 26799652 Free PMC article. No abstract available.
-
Seeing is believing: methods to monitor vertebrate autophagy in vivo.Open Biol. 2018 Oct 24;8(10):180106. doi: 10.1098/rsob.180106. Open Biol. 2018. PMID: 30355753 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
