

Synchrotron-based small-angle X-ray scattering of proteins in solution
- PMID: 24967622
- PMCID: PMC4472361
- DOI: 10.1038/nprot.2014.116
Synchrotron-based small-angle X-ray scattering of proteins in solution
Abstract
With recent advances in data analysis algorithms, X-ray detectors and synchrotron sources, small-angle X-ray scattering (SAXS) has become much more accessible to the structural biology community. Although limited to ∼10 Å resolution, SAXS can provide a wealth of structural information on biomolecules in solution and is compatible with a wide range of experimental conditions. SAXS is thus an attractive alternative when crystallography is not possible. Moreover, advanced use of SAXS can provide unique insight into biomolecular behavior that can only be observed in solution, such as large conformational changes and transient protein-protein interactions. Unlike crystal diffraction data, however, solution scattering data are subtle in appearance, highly sensitive to sample quality and experimental errors and easily misinterpreted. In addition, synchrotron beamlines that are dedicated to SAXS are often unfamiliar to the nonspecialist. Here we present a series of procedures that can be used for SAXS data collection and basic cross-checks designed to detect and avoid aggregation, concentration effects, radiation damage, buffer mismatch and other common problems. Human serum albumin (HSA) serves as a convenient and easily replicated example of just how subtle these problems can sometimes be, but also of how proper technique can yield pristine data even in problematic cases. Because typical data collection times at a synchrotron are only one to several days, we recommend that the sample purity, homogeneity and solubility be extensively optimized before the experiment.
Conflict of interest statement
Figures

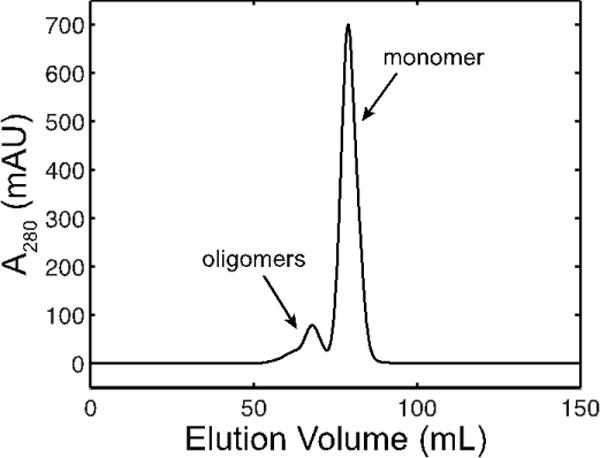
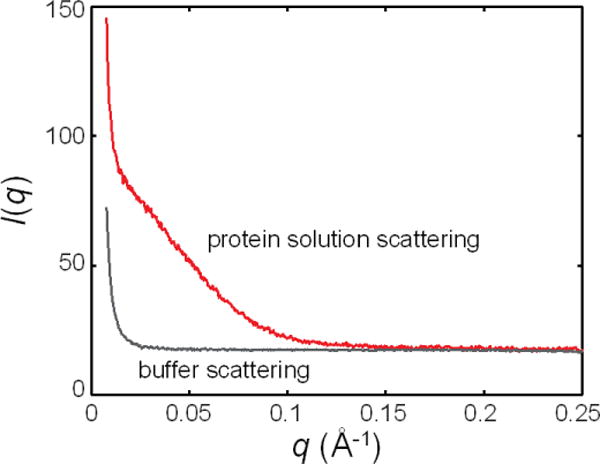
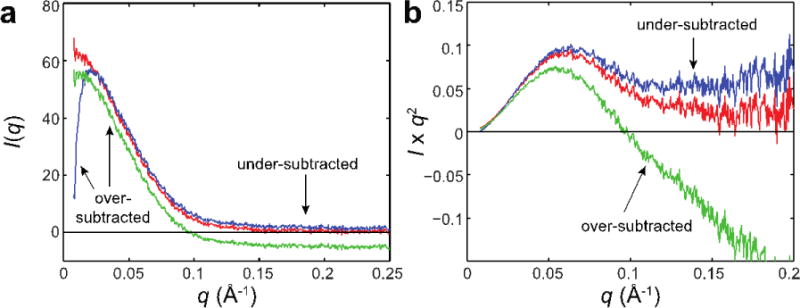
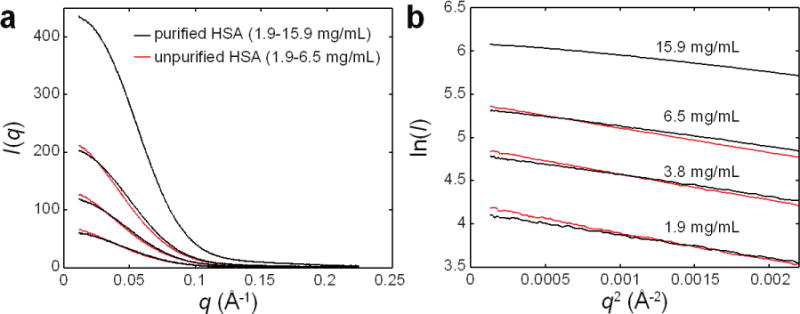
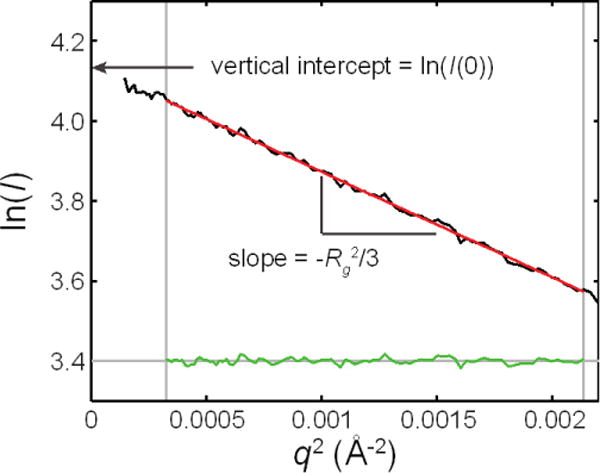
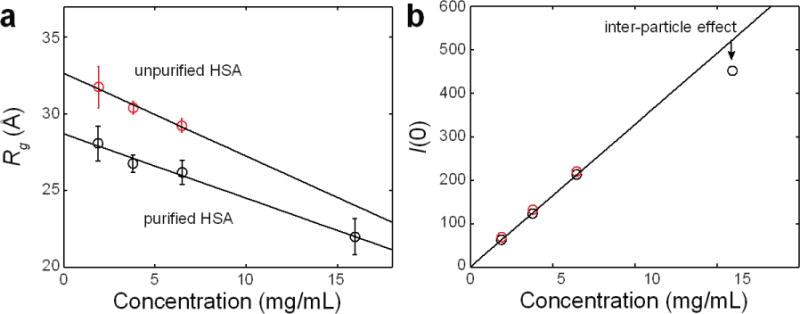
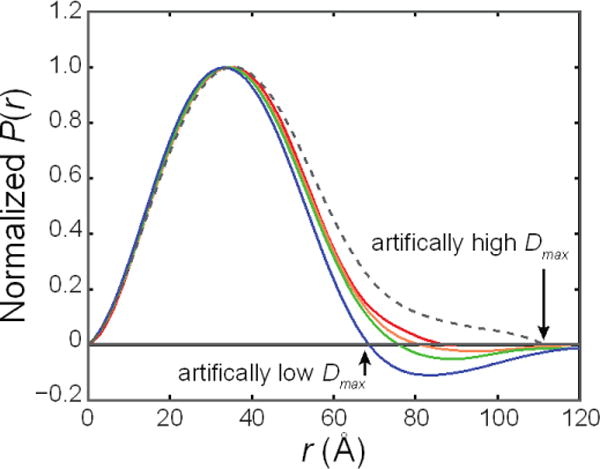

References
-
- Nagar B, Kuriyan J. SAXS and the working protein. Structure. 2005;13:169–170. - PubMed
-
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
