

Library preparation for highly accurate population sequencing of RNA viruses
- PMID: 24967624
- PMCID: PMC4418788
- DOI: 10.1038/nprot.2014.118
Library preparation for highly accurate population sequencing of RNA viruses
Abstract
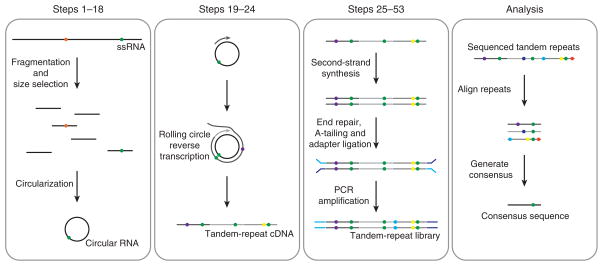
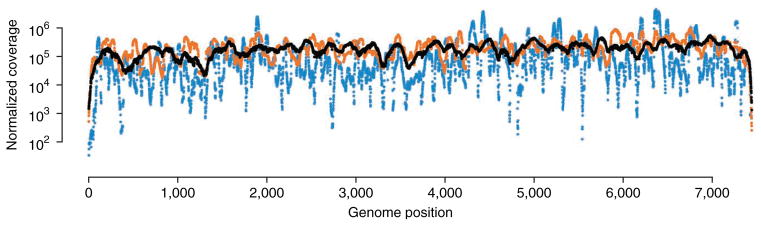
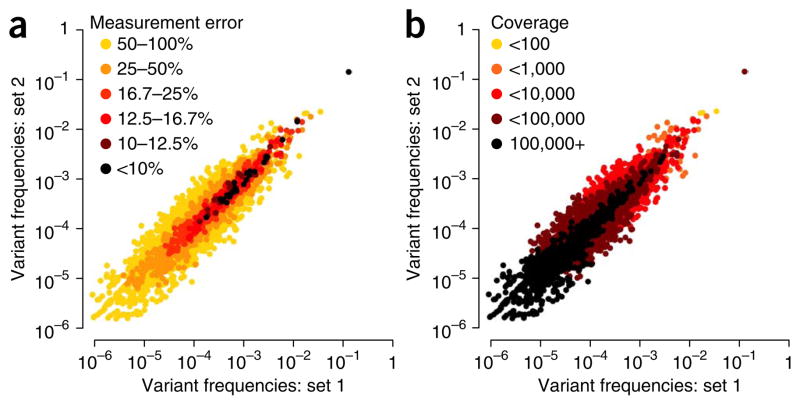
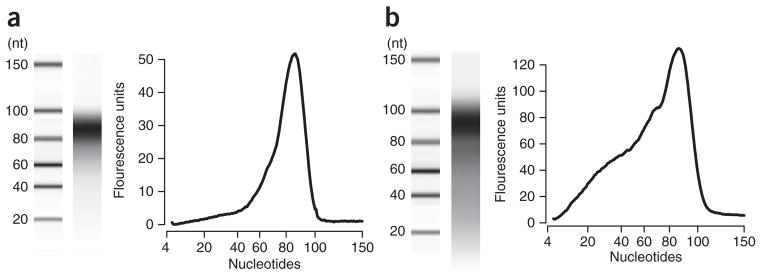
Circular resequencing (CirSeq) is a novel technique for efficient and highly accurate next-generation sequencing (NGS) of RNA virus populations. The foundation of this approach is the circularization of fragmented viral RNAs, which are then redundantly encoded into tandem repeats by 'rolling-circle' reverse transcription. When sequenced, the redundant copies within each read are aligned to derive a consensus sequence of their initial RNA template. This process yields sequencing data with error rates far below the variant frequencies observed for RNA viruses, facilitating ultra-rare variant detection and accurate measurement of low-frequency variants. Although library preparation takes ∼5 d, the high-quality data generated by CirSeq simplifies downstream data analysis, making this approach substantially more tractable for experimentalists.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
