

Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion
- PMID: 24967968
- PMCID: PMC4079937
- DOI: 10.1038/cddis.2014.272
Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion
Abstract
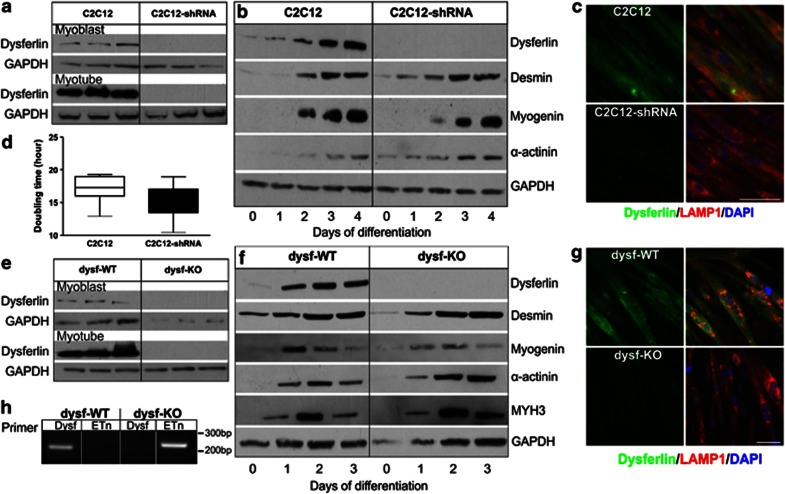
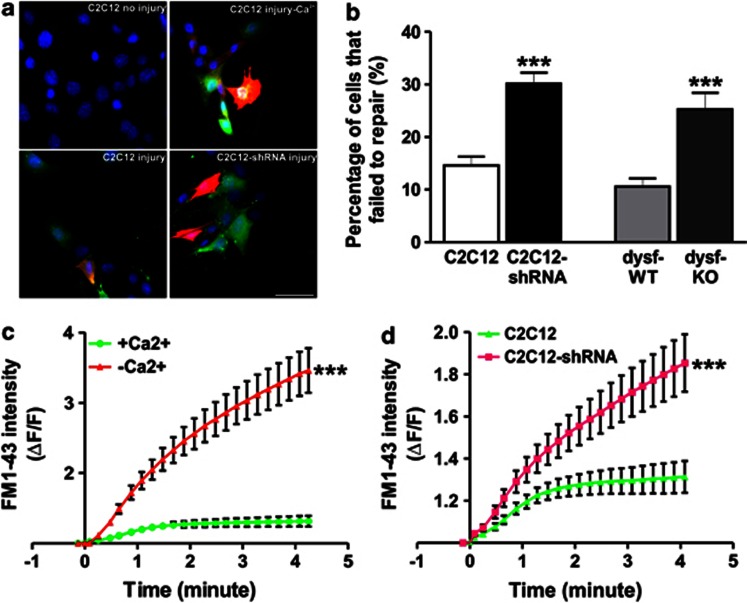
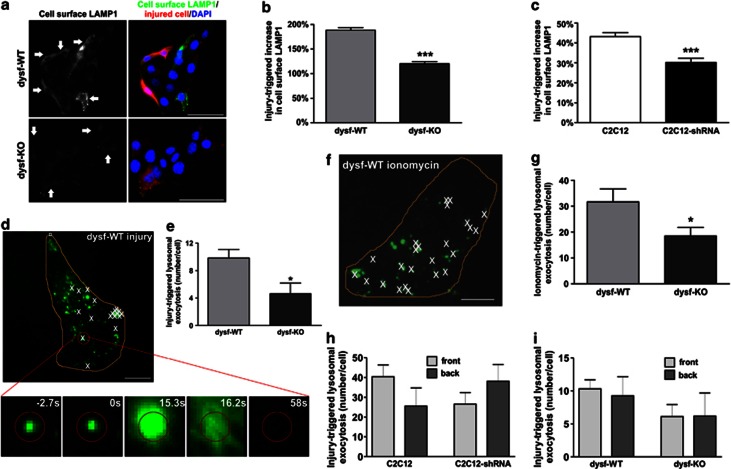
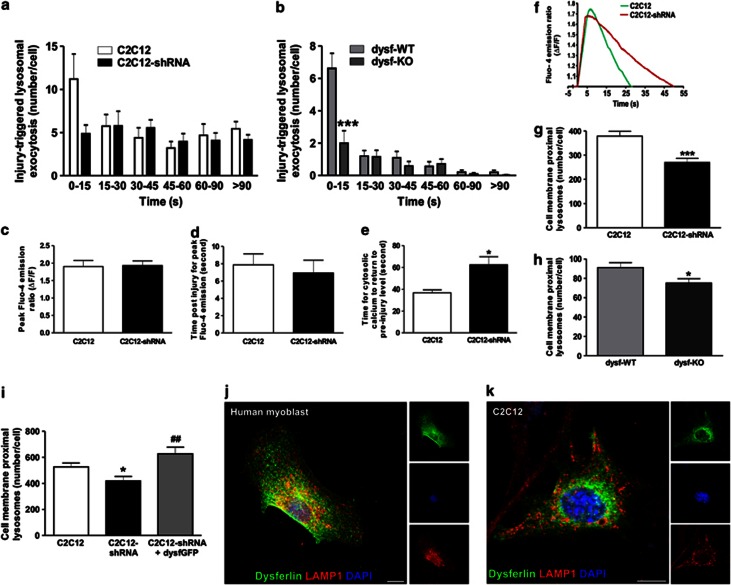
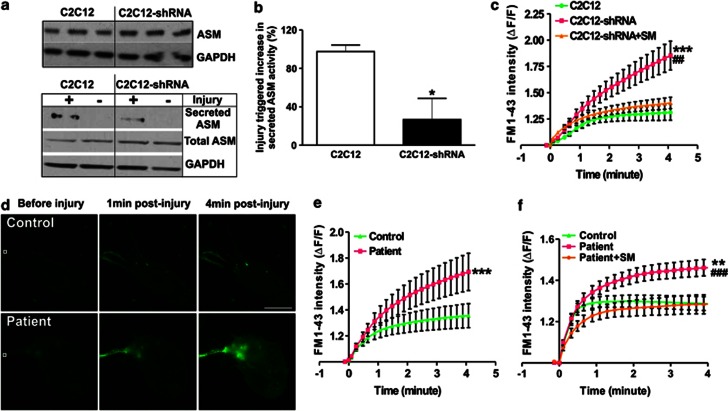
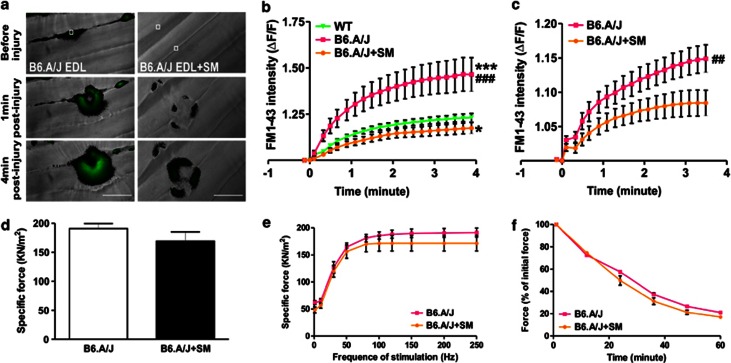
Dysferlin deficiency compromises the repair of injured muscle, but the underlying cellular mechanism remains elusive. To study this phenomenon, we have developed mouse and human myoblast models for dysferlinopathy. These dysferlinopathic myoblasts undergo normal differentiation but have a deficit in their ability to repair focal injury to their cell membrane. Imaging cells undergoing repair showed that dysferlin-deficit decreased the number of lysosomes present at the cell membrane, resulting in a delay and reduction in injury-triggered lysosomal exocytosis. We find repair of injured cells does not involve formation of intracellular membrane patch through lysosome-lysosome fusion; instead, individual lysosomes fuse with the injured cell membrane, releasing acid sphingomyelinase (ASM). ASM secretion was reduced in injured dysferlinopathic cells, and acute treatment with sphingomyelinase restored the repair ability of dysferlinopathic myoblasts and myofibers. Our results provide the mechanism for dysferlin-mediated repair of skeletal muscle sarcolemma and identify ASM as a potential therapy for dysferlinopathy.
Figures
References
-
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet 1998; 20: 37–42. - PubMed
-
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998; 20: 31–36. - PubMed
-
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003; 423: 168–172. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
