

Endothelial dysfunction in chronic inflammatory diseases
- PMID: 24968272
- PMCID: PMC4139785
- DOI: 10.3390/ijms150711324
Endothelial dysfunction in chronic inflammatory diseases
Abstract
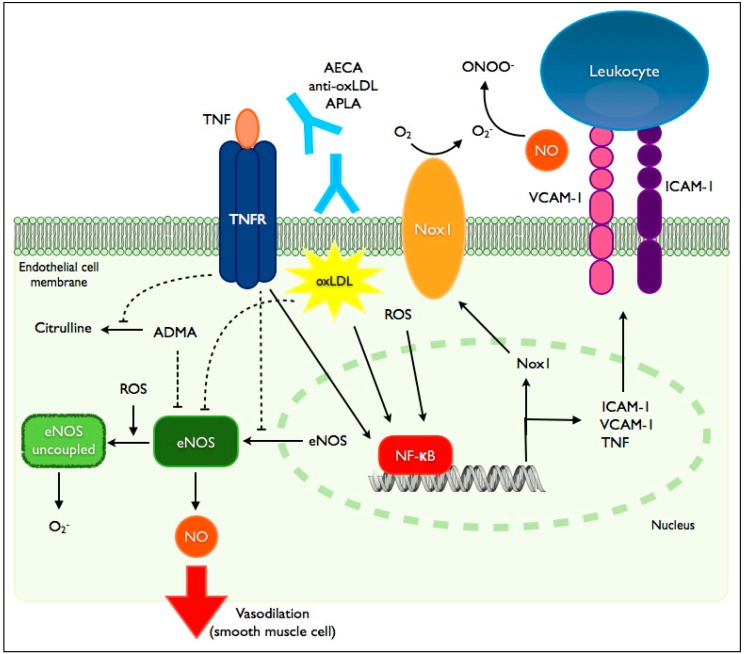
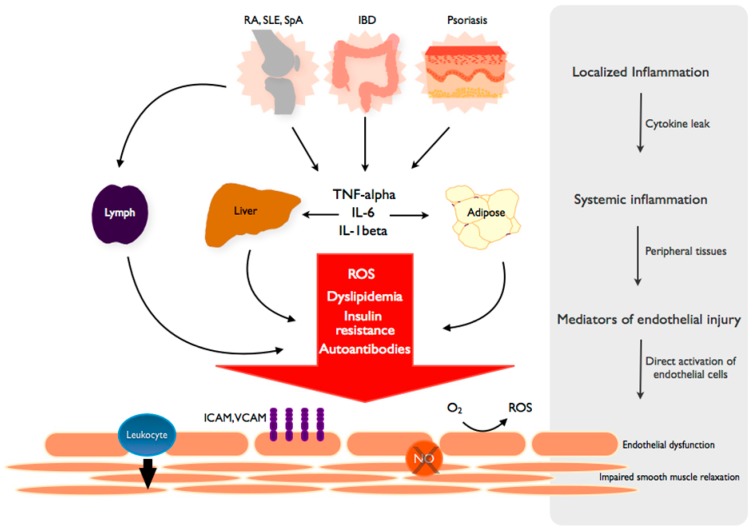
Chronic inflammatory diseases are associated with accelerated atherosclerosis and increased risk of cardiovascular diseases (CVD). As the pathogenesis of atherosclerosis is increasingly recognized as an inflammatory process, similarities between atherosclerosis and systemic inflammatory diseases such as rheumatoid arthritis, inflammatory bowel diseases, lupus, psoriasis, spondyloarthritis and others have become a topic of interest. Endothelial dysfunction represents a key step in the initiation and maintenance of atherosclerosis and may serve as a marker for future risk of cardiovascular events. Patients with chronic inflammatory diseases manifest endothelial dysfunction, often early in the course of the disease. Therefore, mechanisms linking systemic inflammatory diseases and atherosclerosis may be best understood at the level of the endothelium. Multiple factors, including circulating inflammatory cytokines, TNF-α (tumor necrosis factor-α), reactive oxygen species, oxidized LDL (low density lipoprotein), autoantibodies and traditional risk factors directly and indirectly activate endothelial cells, leading to impaired vascular relaxation, increased leukocyte adhesion, increased endothelial permeability and generation of a pro-thrombotic state. Pharmacologic agents directed against TNF-α-mediated inflammation may decrease the risk of endothelial dysfunction and cardiovascular disease in these patients. Understanding the precise mechanisms driving endothelial dysfunction in patients with systemic inflammatory diseases may help elucidate the pathogenesis of atherosclerosis in the general population.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
