

Background selection as baseline for nucleotide variation across the Drosophila genome
- PMID: 24968283
- PMCID: PMC4072542
- DOI: 10.1371/journal.pgen.1004434
Background selection as baseline for nucleotide variation across the Drosophila genome
Abstract
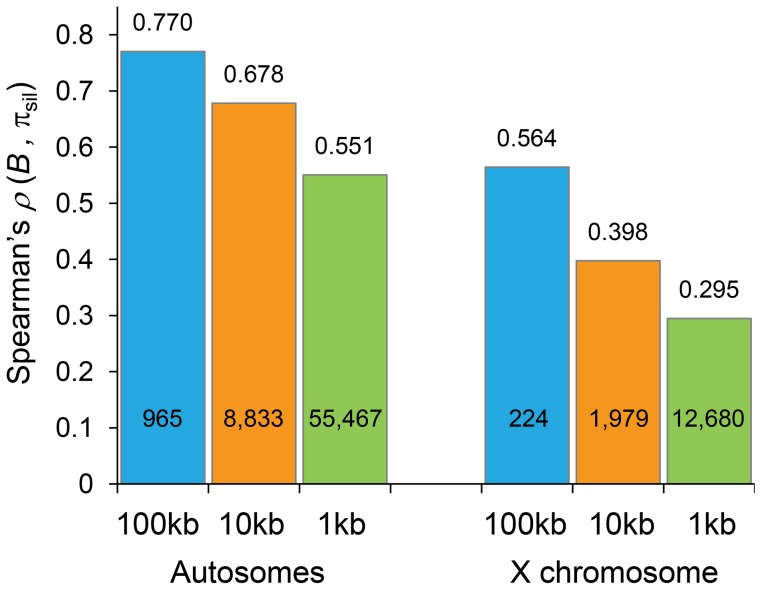
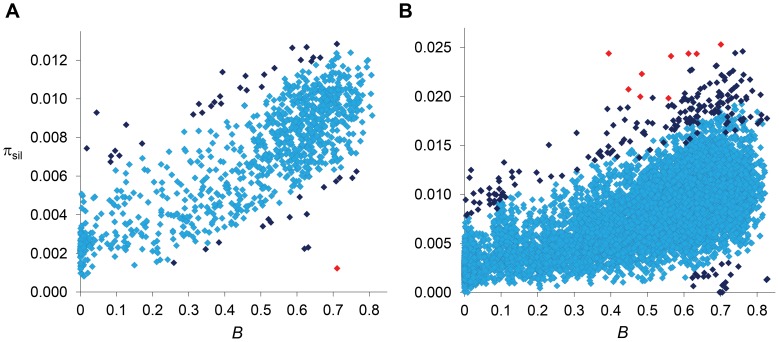
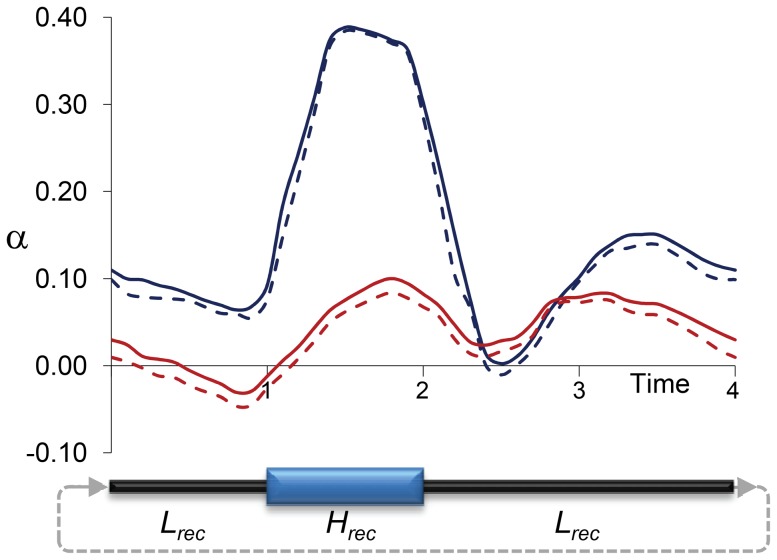
The constant removal of deleterious mutations by natural selection causes a reduction in neutral diversity and efficacy of selection at genetically linked sites (a process called Background Selection, BGS). Population genetic studies, however, often ignore BGS effects when investigating demographic events or the presence of other types of selection. To obtain a more realistic evolutionary expectation that incorporates the unavoidable consequences of deleterious mutations, we generated high-resolution landscapes of variation across the Drosophila melanogaster genome under a BGS scenario independent of polymorphism data. We find that BGS plays a significant role in shaping levels of variation across the entire genome, including long introns and intergenic regions distant from annotated genes. We also find that a very large percentage of the observed variation in diversity across autosomes can be explained by BGS alone, up to 70% across individual chromosome arms at 100-kb scale, thus indicating that BGS predictions can be used as baseline to infer additional types of selection and demographic events. This approach allows detecting several outlier regions with signal of recent adaptive events and selective sweeps. The use of a BGS baseline, however, is particularly appropriate to investigate the presence of balancing selection and our study exposes numerous genomic regions with the predicted signature of higher polymorphism than expected when a BGS context is taken into account. Importantly, we show that these conclusions are robust to the mutation and selection parameters of the BGS model. Finally, analyses of protein evolution together with previous comparisons of genetic maps between Drosophila species, suggest temporally variable recombination landscapes and, thus, local BGS effects that may differ between extant and past phases. Because genome-wide BGS and temporal changes in linkage effects can skew approaches to estimate demographic and selective events, future analyses should incorporate BGS predictions and capture local recombination variation across genomes and along lineages.
Conflict of interest statement
The author has declared that no competing interests exist.
Figures
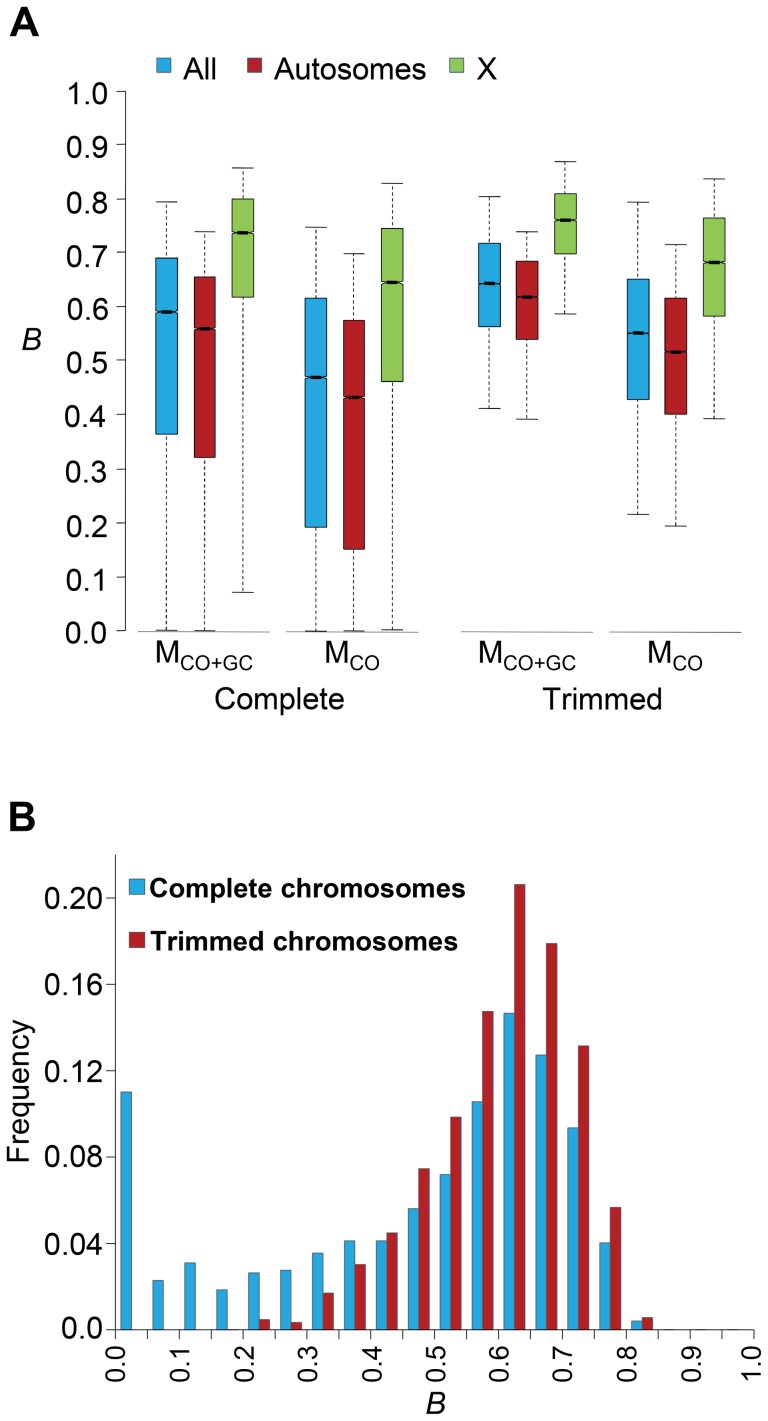
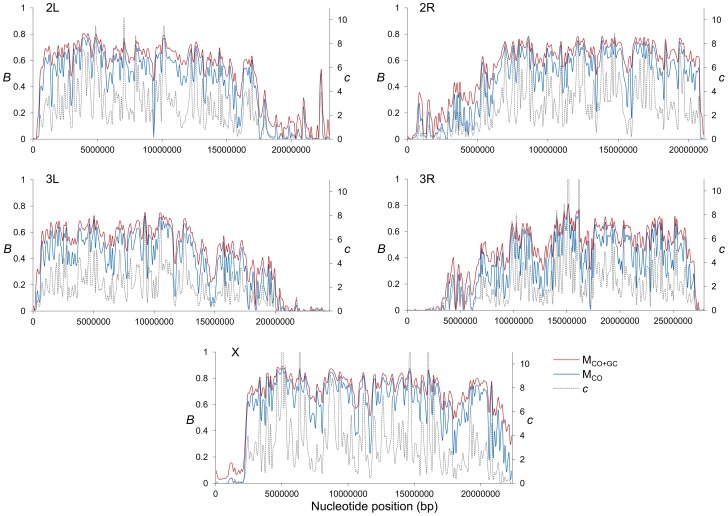
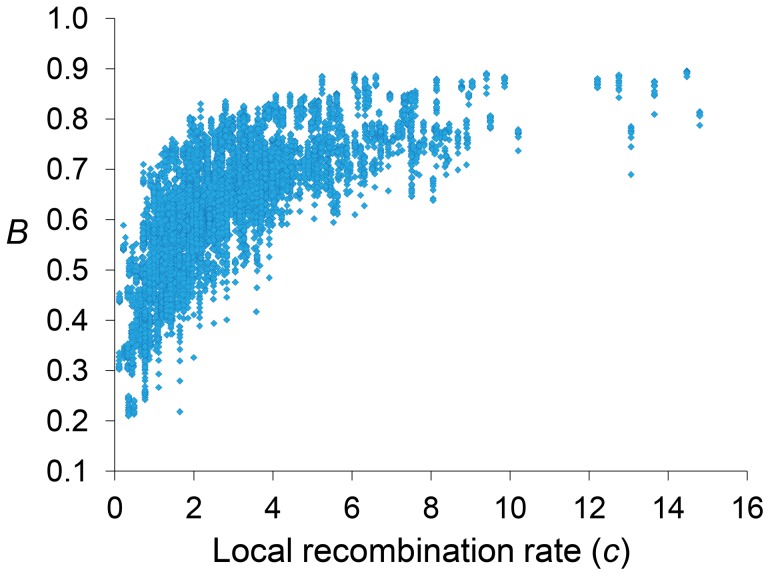
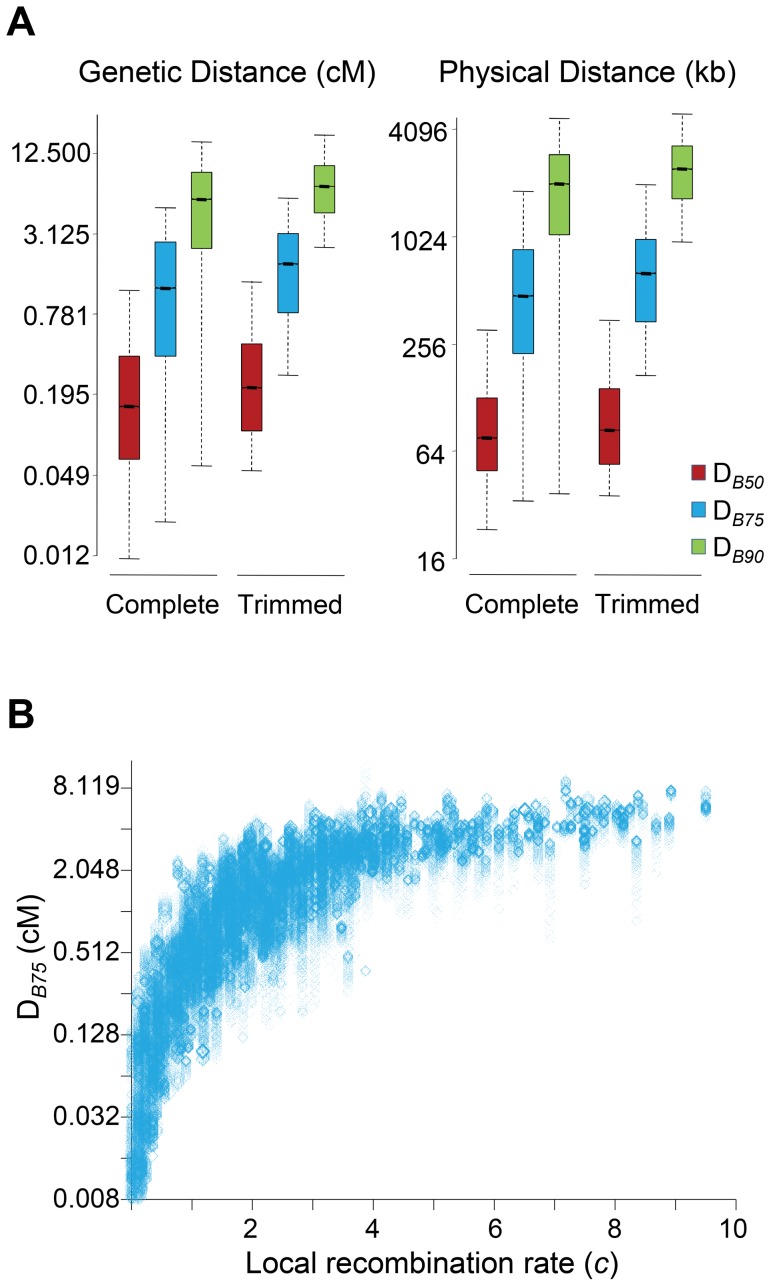
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
