

An integrated computational approach can classify VHL missense mutations according to risk of clear cell renal carcinoma
- PMID: 24969085
- PMCID: PMC4204774
- DOI: 10.1093/hmg/ddu321
An integrated computational approach can classify VHL missense mutations according to risk of clear cell renal carcinoma
Abstract
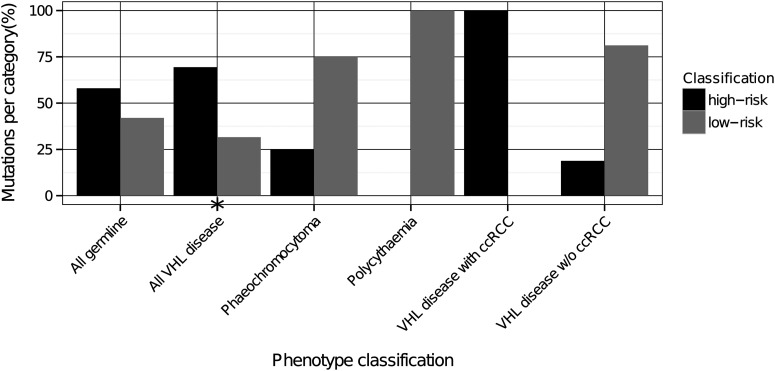
Mutations in the von Hippel-Lindau (VHL) gene are pathogenic in VHL disease, congenital polycythaemia and clear cell renal carcinoma (ccRCC). pVHL forms a ternary complex with elongin C and elongin B, critical for pVHL stability and function, which interacts with Cullin-2 and RING-box protein 1 to target hypoxia-inducible factor for polyubiquitination and proteasomal degradation. We describe a comprehensive database of missense VHL mutations linked to experimental and clinical data. We use predictions from in silico tools to link the functional effects of missense VHL mutations to phenotype. The risk of ccRCC in VHL disease is linked to the degree of destabilization resulting from missense mutations. An optimized binary classification system (symphony), which integrates predictions from five in silico methods, can predict the risk of ccRCC associated with VHL missense mutations with high sensitivity and specificity. We use symphony to generate predictions for risk of ccRCC for all possible VHL missense mutations and present these predictions, in association with clinical and experimental data, in a publically available, searchable web server.
© The Author 2014. Published by Oxford University Press.
Figures





References
-
- Nordstrom-O'Brien M., van der Luijt R.B., van Rooijen E., van den Ouweland A.M., Majoor-Krakauer D.F., Lolkema M.P., van Brussel A., Voest E.E., Giles R.H. Genetic analysis of von Hippel-Lindau disease. Hum. Mutat. 2010;31:521–537. - PubMed
-
- Ang S.O., Chen H., Hirota K., Gordeuk V.R., Jelinek J., Guan Y., Liu E., Sergueeva A.I., Miasnikova G.Y., Mole D., et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002;32:614–621. - PubMed
-
- Pastore Y.D., Jelinek J., Ang S., Guan Y., Liu E., Jedlickova K., Krishnamurti L., Prchal J.T. Mutations in the VHL gene in sporadic apparently congenital polycythemia. Blood. 2003;101:1591–1595. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
