

Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier
- PMID: 24970096
- PMCID: PMC4107747
- DOI: 10.1093/brain/awu165
Pure and syndromic optic atrophy explained by deep intronic OPA1 mutations and an intralocus modifier
Abstract
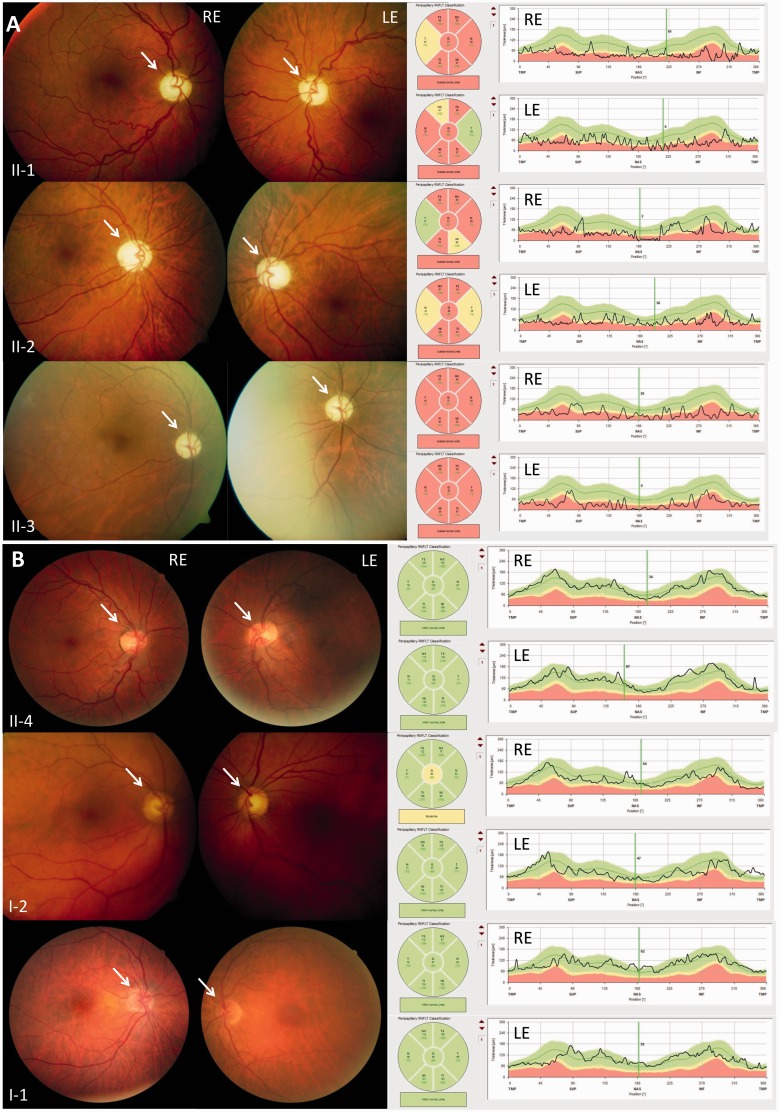
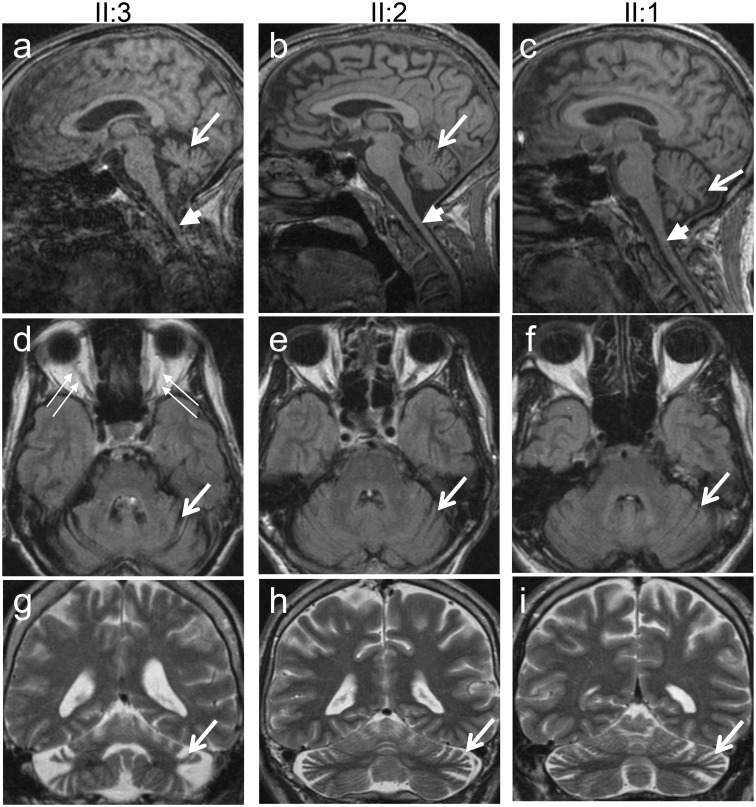
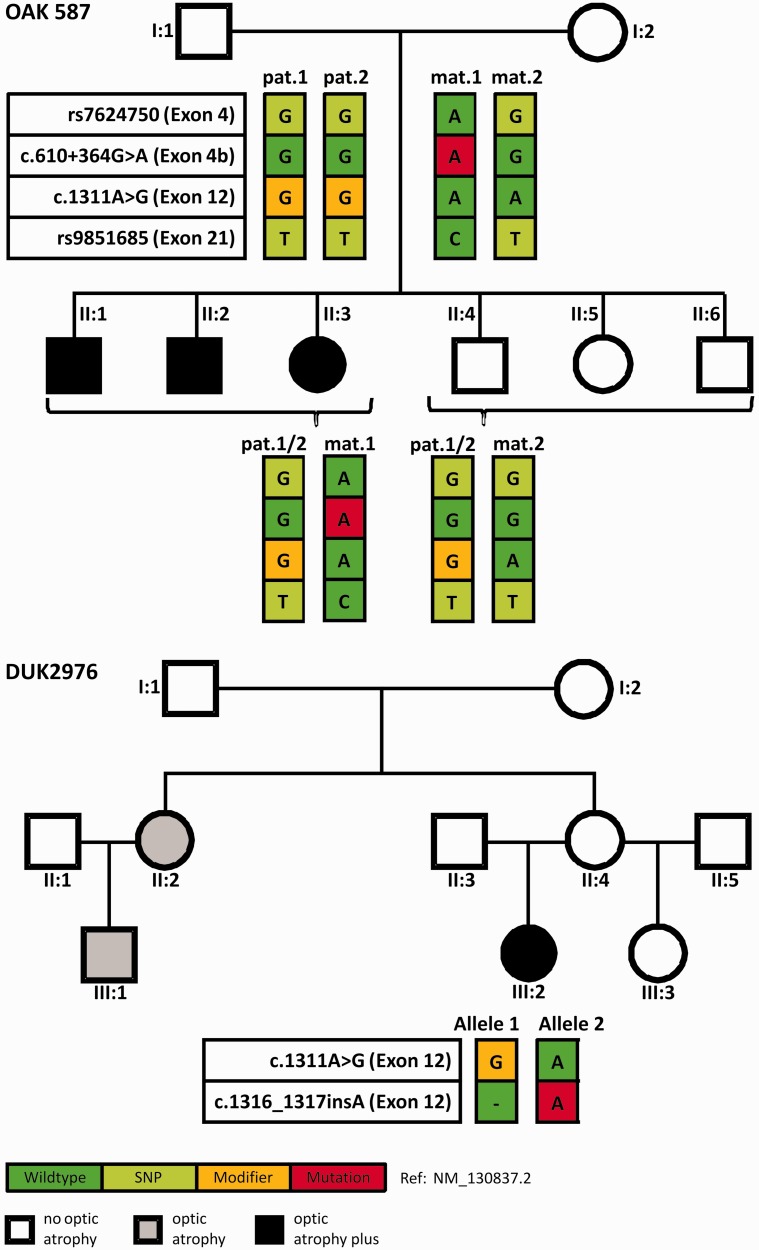
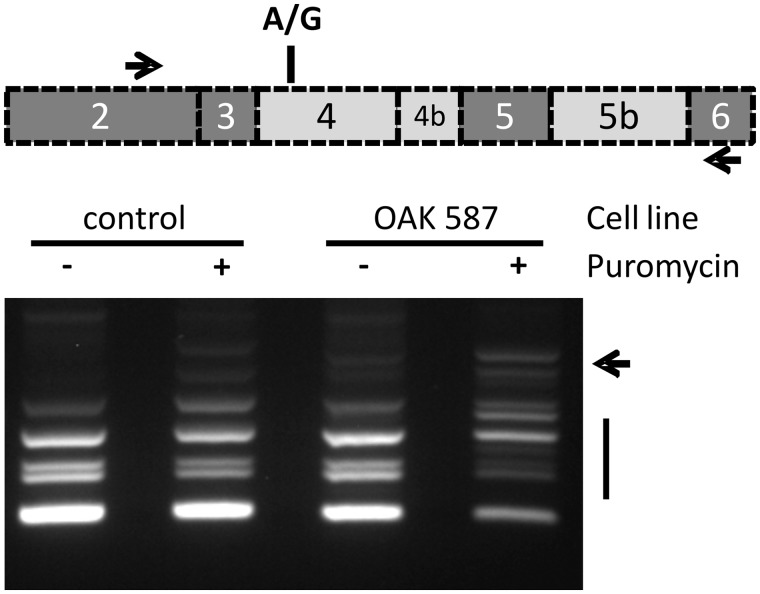
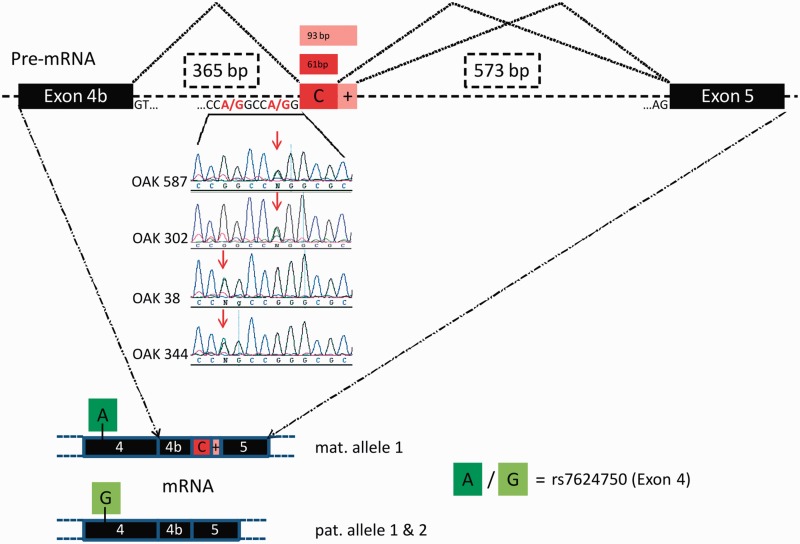
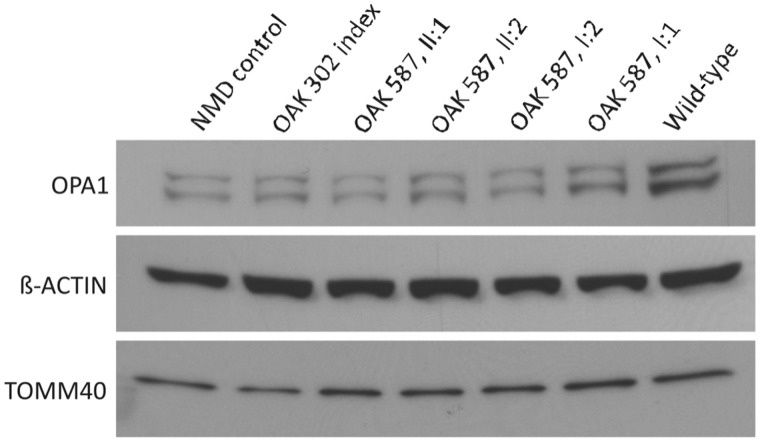
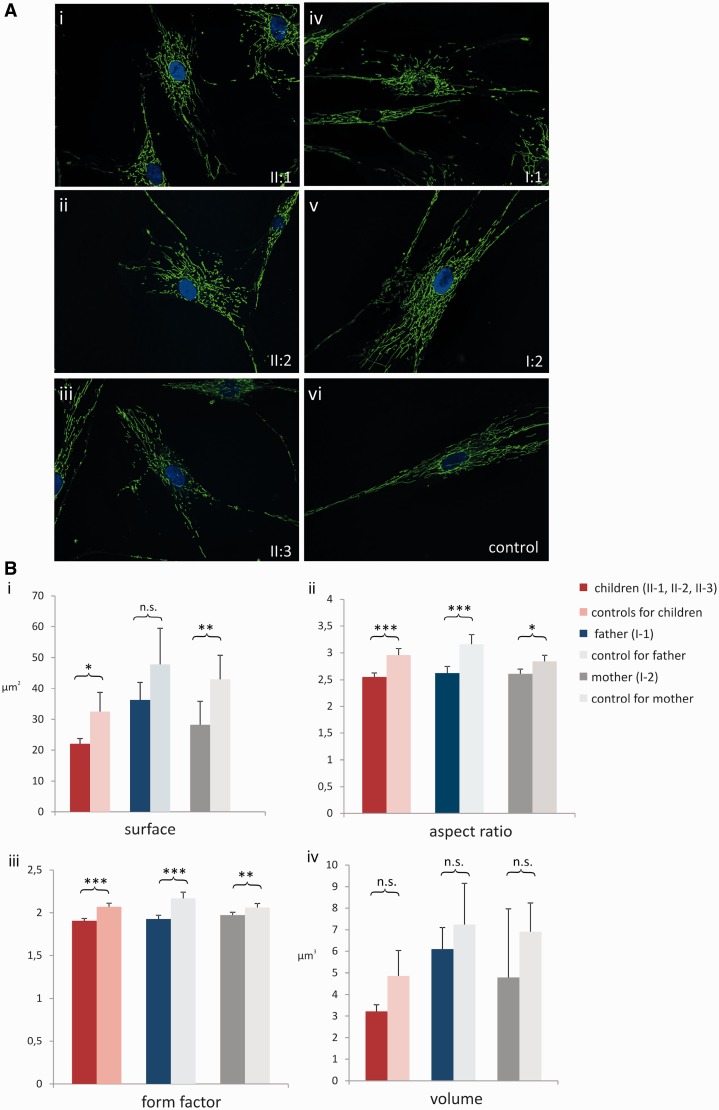
The genetic diagnosis in inherited optic neuropathies often remains challenging, and the emergence of complex neurological phenotypes that involve optic neuropathy is puzzling. Here we unravel two novel principles of genetic mechanisms in optic neuropathies: deep intronic OPA1 mutations, which explain the disease in several so far unsolved cases; and an intralocus OPA1 modifier, which explains the emergence of syndromic 'optic atrophy plus' phenotypes in several families. First, we unravelled a deep intronic mutation 364 base pairs 3' of exon 4b in OPA1 by in-depth investigation of a family with severe optic atrophy plus syndrome in which conventional OPA1 diagnostics including gene dosage analyses were normal. The mutation creates a new splice acceptor site resulting in aberrant OPA1 transcripts with retained intronic sequence and subsequent translational frameshift as shown by complementary DNA analysis. In patient fibroblasts we demonstrate nonsense mediated messenger RNA decay, reduced levels of OPA1 protein, and impairment of mitochondrial dynamics. Subsequent site-specific screening of >360 subjects with unexplained inherited optic neuropathy revealed three additional families carrying this deep intronic mutation and a base exchange four nucleotides upstream, respectively, thus confirming the clinical significance of this mutational mechanism. Second, in all severely affected patients of the index family, the deep intronic mutation occurred in compound heterozygous state with an exonic OPA1 missense variant (p.I382M; NM_015560.2). The variant alone did not cause a phenotype, even in homozygous state indicating that this long debated OPA1 variant is not pathogenic per se, but acts as a phenotypic modifier if it encounters in trans with an OPA1 mutation. Subsequent screening of whole exomes from >600 index patients identified a second family with severe optic atrophy plus syndrome due to compound heterozygous p.I382M, thus confirming this mechanism. In summary, we provide genetic and functional evidence that deep intronic mutations in OPA1 can cause optic atrophy and explain disease in a substantial share of families with unsolved inherited optic neuropathies. Moreover, we show that an OPA1 modifier variant explains the emergence of optic atrophy plus phenotypes if combined in trans with another OPA1 mutation. Both mutational mechanisms identified in this study-deep intronic mutations and intragenic modifiers-might represent more generalizable mechanisms that could be found also in a wide range of other neurodegenerative and optic neuropathy diseases.
Keywords: ataxia; cryptic exon; deep intronic mutation; genetic modifier; mitochondrial network.
© The Author (2014). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Mutation survey of the optic atrophy 1 gene in 193 Chinese families with suspected hereditary optic neuropathy.Mol Vis. 2013;19:292-302. Epub 2013 Feb 6. Mol Vis. 2013. PMID: 23401657 Free PMC article.
-
Characterization of two novel intronic OPA1 mutations resulting in aberrant pre-mRNA splicing.BMC Med Genet. 2017 Feb 28;18(1):22. doi: 10.1186/s12881-017-0383-x. BMC Med Genet. 2017. PMID: 28245802 Free PMC article.
-
A comprehensive survey of mutations in the OPA1 gene in patients with autosomal dominant optic atrophy.Invest Ophthalmol Vis Sci. 2002 Jun;43(6):1715-24. Invest Ophthalmol Vis Sci. 2002. PMID: 12036970
-
Recessive optic atrophy, sensorimotor neuropathy and cataract associated with novel compound heterozygous mutations in OPA1.Mol Med Rep. 2016 Jul;14(1):33-40. doi: 10.3892/mmr.2016.5209. Epub 2016 May 4. Mol Med Rep. 2016. PMID: 27150940 Free PMC article. Review.
-
Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy.Mitochondrion. 2019 May;46:262-269. doi: 10.1016/j.mito.2018.07.006. Epub 2018 Aug 27. Mitochondrion. 2019. PMID: 30165240
Cited by
-
CADD score has limited clinical validity for the identification of pathogenic variants in noncoding regions in a hereditary cancer panel.Genet Med. 2016 Dec;18(12):1269-1275. doi: 10.1038/gim.2016.44. Epub 2016 May 5. Genet Med. 2016. PMID: 27148939 Free PMC article.
-
Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders.Eur J Hum Genet. 2016 Oct;24(10):1460-6. doi: 10.1038/ejhg.2016.42. Epub 2016 May 11. Eur J Hum Genet. 2016. PMID: 27165006 Free PMC article.
-
Saccharomyces cerevisiae as a Tool for Studying Mutations in Nuclear Genes Involved in Diseases Caused by Mitochondrial DNA Instability.Genes (Basel). 2021 Nov 24;12(12):1866. doi: 10.3390/genes12121866. Genes (Basel). 2021. PMID: 34946817 Free PMC article. Review.
-
Metabolic stroke in a patient with bi-allelic OPA1 mutations.Metab Brain Dis. 2019 Aug;34(4):1043-1048. doi: 10.1007/s11011-019-00415-2. Epub 2019 Apr 10. Metab Brain Dis. 2019. PMID: 30972688
-
Metabolomics hallmarks OPA1 variants correlating with their in vitro phenotype and predicting clinical severity.Hum Mol Genet. 2020 May 28;29(8):1319-1329. doi: 10.1093/hmg/ddaa047. Hum Mol Genet. 2020. PMID: 32202296 Free PMC article.
References
-
- Amati-Bonneau P, Guichet A, Olichon A, Chevrollier A, Viala F, Miot S, et al. OPA1 R445H mutation in optic atrophy associated with sensorineural deafness. Ann Neurol. 2005;58:958–63. - PubMed
-
- Amati-Bonneau P, Valentino ML, Reynier P, Gallardo ME, Bornstein B, Boissiere A, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–51. - PubMed
-
- Cohn AC, Toomes C, Potter C, Towns KV, Hewitt AW, Inglehearn CF, et al. Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmol. 2007;143:656–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
