

Decoding F508del misfolding in cystic fibrosis
- PMID: 24970227
- PMCID: PMC4101494
- DOI: 10.3390/biom4020498
Decoding F508del misfolding in cystic fibrosis
Abstract
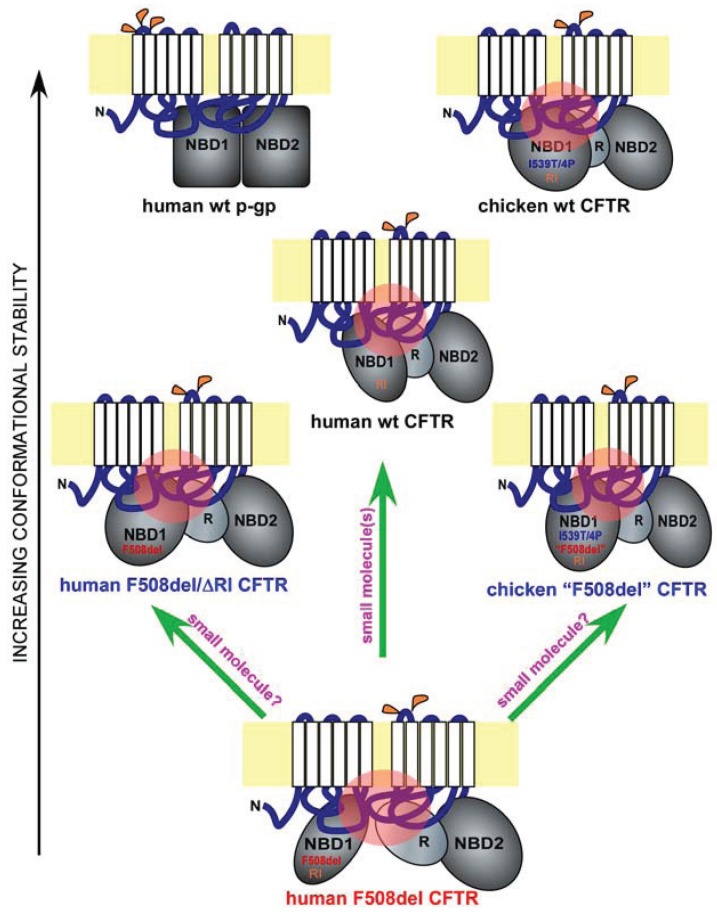
The functional deficiency of the cystic fibrosis transmembrane conductance regulator (CFTR), a plasma membrane chloride channel, leads to the development of cystic fibrosis. The deletion of a phenylalanine at residue 508 (F508del) is the most common cause of CFTR misfolding leading to the disease. The F508del misfolding originates in the first nucleotide-binding domain (NBD1), which induces a global conformational change in CFTR through NBD1's interactions with other domains. Such global misfolding produces a mutant chloride channel that is impaired in exocytic trafficking, peripheral stability, and channel gating. The nature and atomic details of F508del misfolding have been subject to extensive research during the past decade. Current data support a central role for NBD1 in F508del misfolding and rescue. Many cis-acting NBD1 second-site mutations rescue F508del misfolding in the context of full-length CFTR. While some of these mutations appear to specifically counteract the F508del-induced misfolding, others release certain inherent conformational constraints of the human wild-type CFTR. Several small-molecule correctors were recently found to act on key interdomain interfaces of F508del CFTR. Potential rational approaches have been proposed in an attempt to develop highly effective small molecule modulators that improve the cell surface functional expression of F508del CFTR.
Figures
References
-
- Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L., et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Cheng S.H., Gregory R.J., Marshall J., Paul S., Souza D.W., White G.A., O’Riordan C.R., Smith A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. - PubMed
-
- Denning G.M., Anderson M.P., Amara J.F., Marshall J., Smith A.E., Welsh M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
