

Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities
- PMID: 24971906
- PMCID: PMC4137396
- DOI: 10.1016/j.taap.2014.06.010
Comparative toxicity and efficacy of engineered anthrax lethal toxin variants with broad anti-tumor activities
Abstract
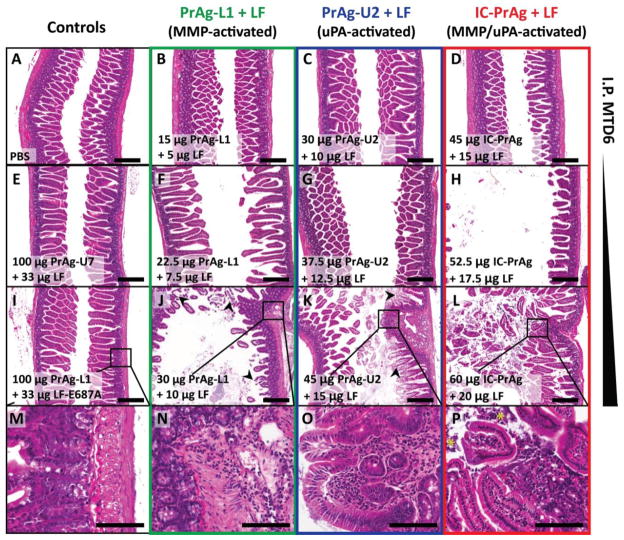
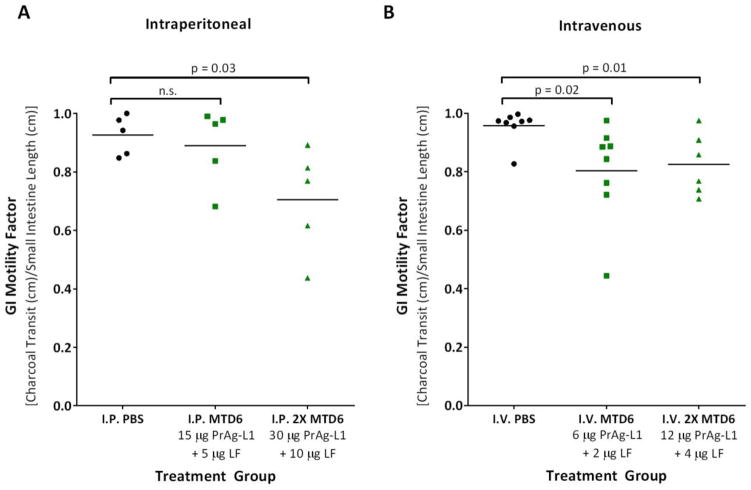
We have previously designed and characterized versions of anthrax lethal toxin that are selectively cytotoxic in the tumor microenvironment and which display broad and potent anti-tumor activities in vivo. Here, we have performed the first direct comparison of the safety and efficacy of three engineered anthrax lethal toxin variants requiring activation by either matrix-metalloproteinases (MMPs), urokinase plasminogen activator (uPA) or co-localized MMP/uPA activities. C57BL/6J mice were challenged with six doses of engineered toxins via intraperitoneal (I.P.) or intravenous (I.V.) dose routes to determine the maximum tolerated dose for six administrations (MTD6) and dose-limiting toxicities. Efficacy was evaluated using the B16-BL6 syngraft model of melanoma; mice bearing established tumors were treated with six I.P. doses of toxin and tumor measurements and immunohistochemistry, paired with terminal blood work, were used to elaborate upon the anti-tumor mechanism and relative efficacy of each variant. We found that MMP-, uPA- and dual MMP/uPA-activated anthrax lethal toxins exhibited the same dose-limiting toxicity; dose-dependent GI toxicity. In terms of efficacy, all three toxins significantly reduced primary B16-BL6 tumor burden, ranging from 32% to 87% reduction, and they also delayed disease progression as evidenced by dose-dependent normalization of blood work values. While target organ toxicity and effective doses were similar amongst the variants, the dual MMP/uPA-activated anthrax lethal toxin exhibited the highest I.P. MTD6 and was 1.5-3-fold better tolerated than the single MMP- and uPA-activated toxins. Overall, we demonstrate that this dual MMP/uPA-activated anthrax lethal toxin can be administered safely and is highly effective in a preclinical model of melanoma. This modified bacterial cytotoxin is thus a promising candidate for further clinical development and evaluation for use in treating human cancers.
Keywords: Bacterial cytotoxin; Cancer; Melanoma; Prodrug; Protease.
Published by Elsevier Inc.
Figures
References
-
- Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72:1–22. - PubMed
-
- Bugge TH. Proteolysis in carcinogenesis. In: Ensley JF, Gutkind JS, Jacob JR, Lippman SM, editors. Head and Neck Cancer. Academic Press; San Diego: 2003. pp. 137–49.
-
- Kontos CK, Scorilas A. Kallikrein-related peptidases (KLKs): a gene family of novel cancer biomarkers. Clin Chem Lab Med. 2012;50:1877–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
