

Synaptic mechanisms underlying cholinergic control of thalamic reticular nucleus neurons
- PMID: 24973413
- PMCID: PMC4215766
- DOI: 10.1113/jphysiol.2014.277376
Synaptic mechanisms underlying cholinergic control of thalamic reticular nucleus neurons
Abstract
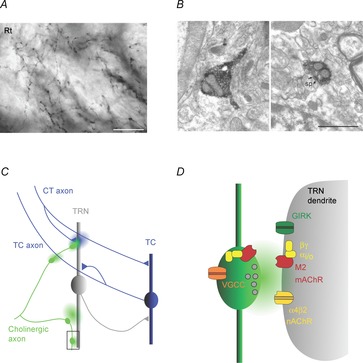
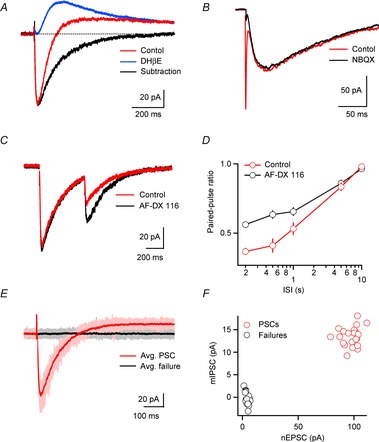
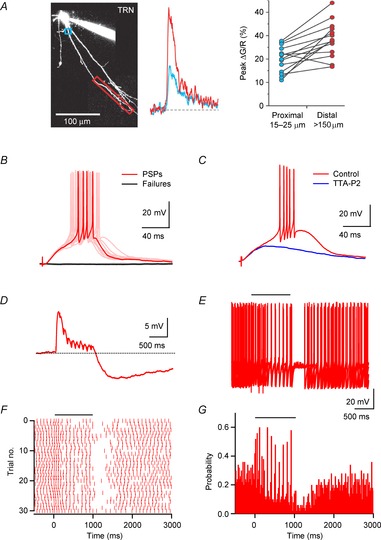
Neuronal networks of the thalamus are the target of extensive cholinergic projections from the basal forebrain and the brainstem. Activation of these afferents can regulate neuronal excitability, transmitter release, and firing patterns in thalamic networks, thereby altering the flow of sensory information during distinct behavioural states. However, cholinergic regulation in the thalamus has been primarily examined by using receptor agonist and antagonist, which has precluded a detailed understanding of the spatiotemporal dynamics that govern cholinergic signalling under physiological conditions. This review summarizes recent studies on cholinergic synaptic transmission in the thalamic reticular nucleus (TRN), a brain structure intimately involved in the control of sensory processing and the generation of rhythmic activity in the thalamocortical system. This work has shown that acetylcholine (ACh) released from individual axons can rapidly and reliably activate both pre- and postsynaptic cholinergic receptors, thereby controlling TRN neuronal activity with high spatiotemporal precision.
© 2014 The Author. The Journal of Physiology © 2014 The Physiological Society.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources