

Degradation of a connexin40 mutant linked to atrial fibrillation is accelerated
- PMID: 24973497
- PMCID: PMC4135452
- DOI: 10.1016/j.yjmcc.2014.06.010
Degradation of a connexin40 mutant linked to atrial fibrillation is accelerated
Abstract
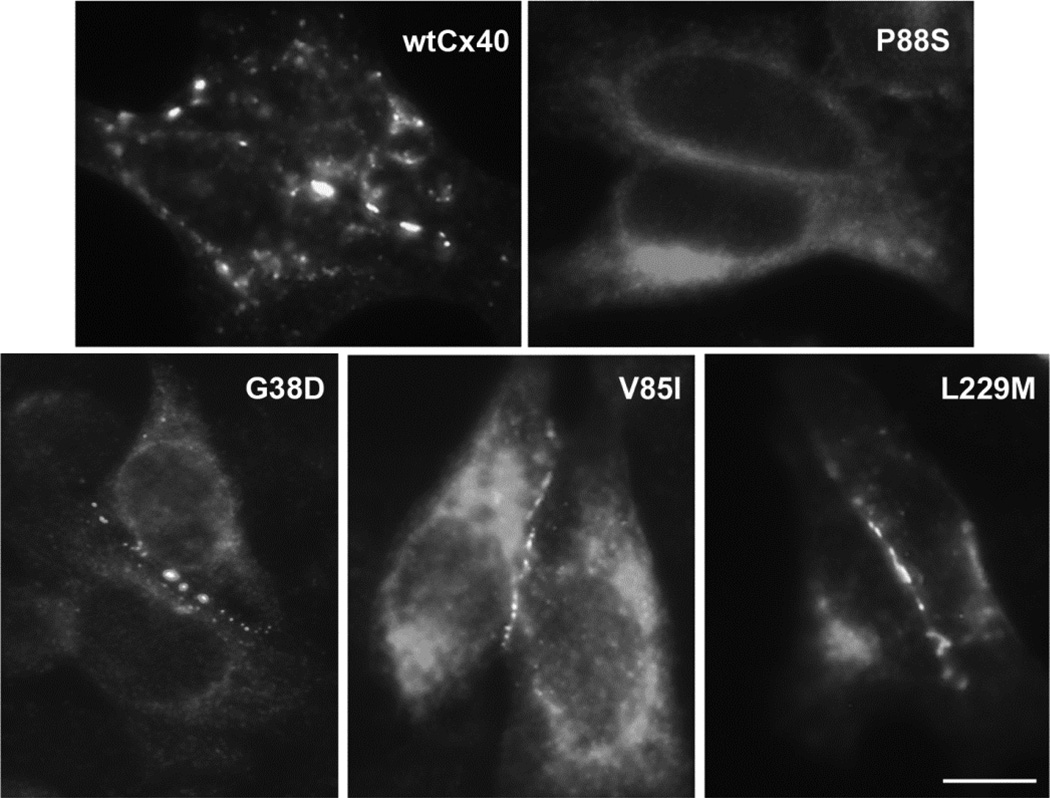
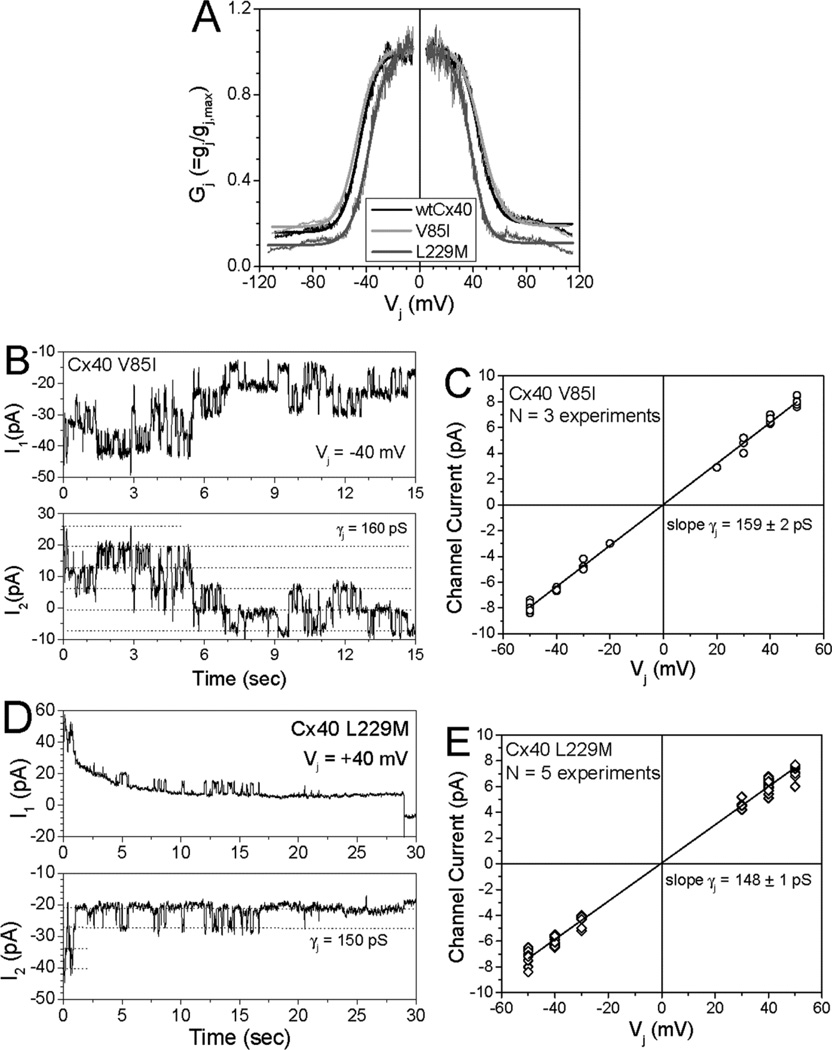
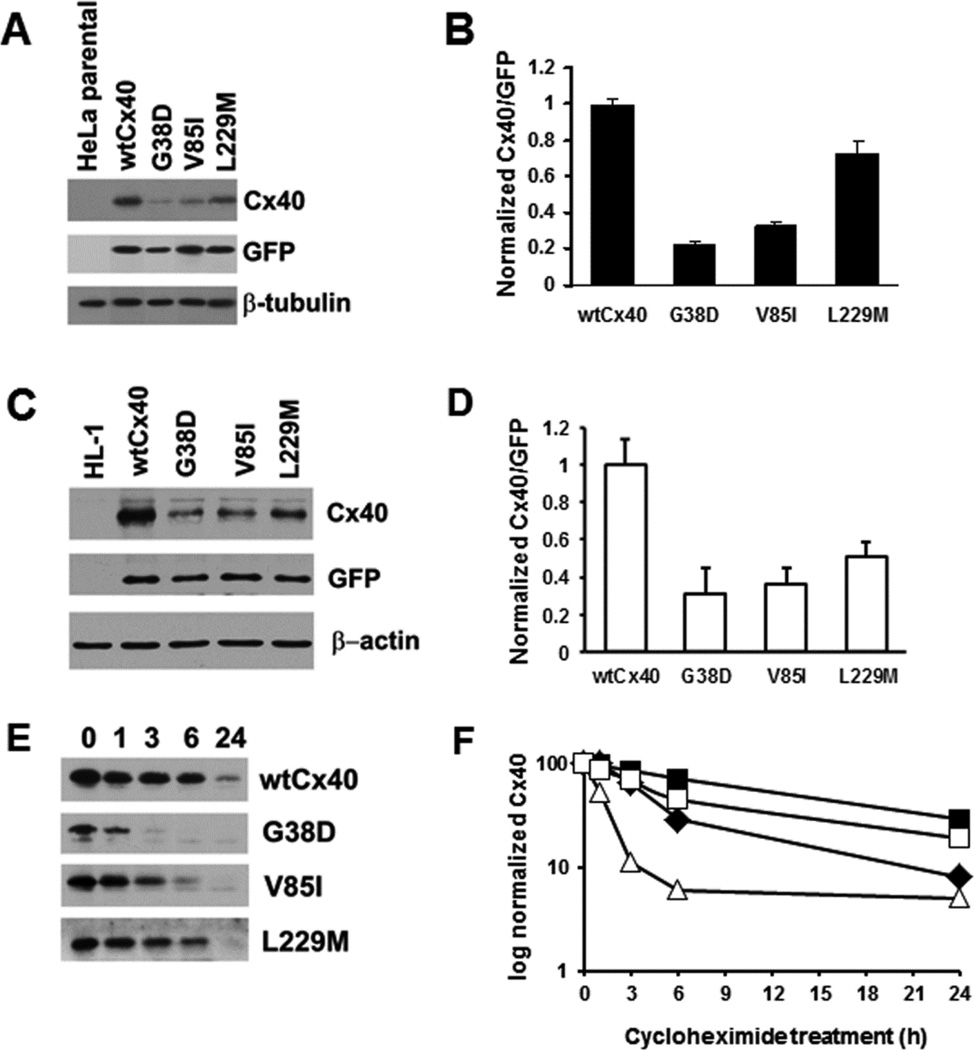
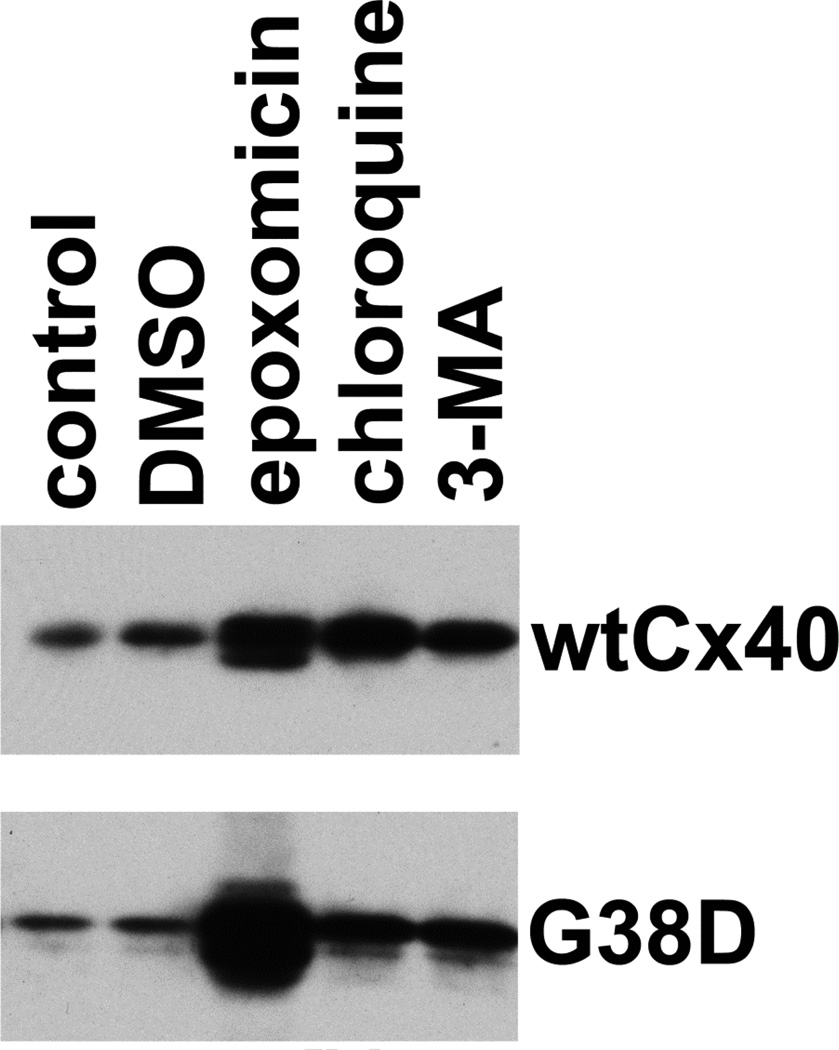
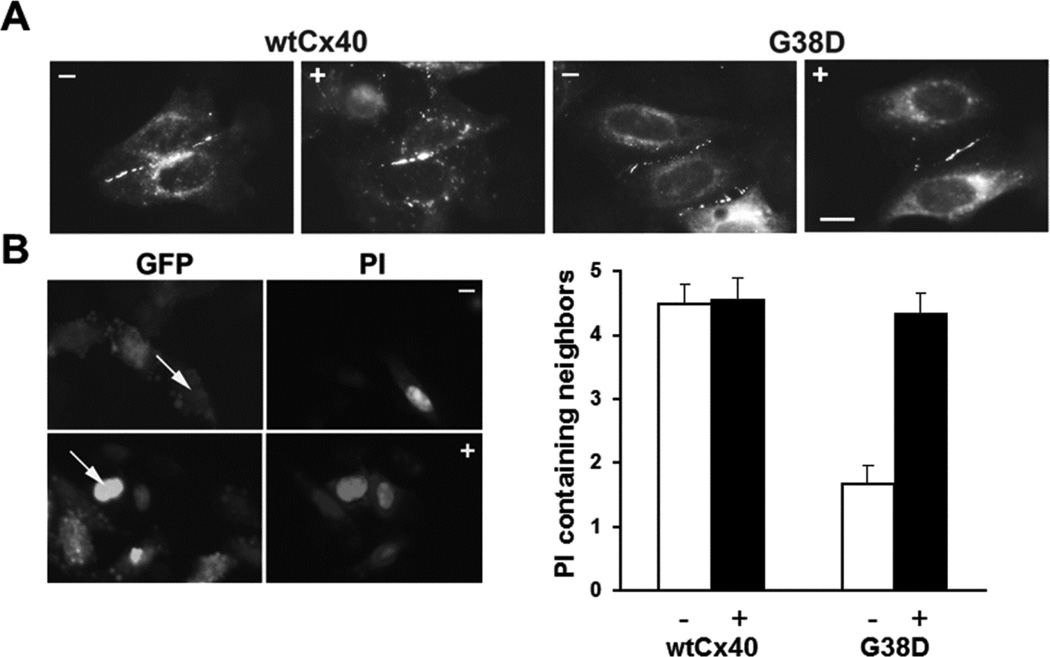
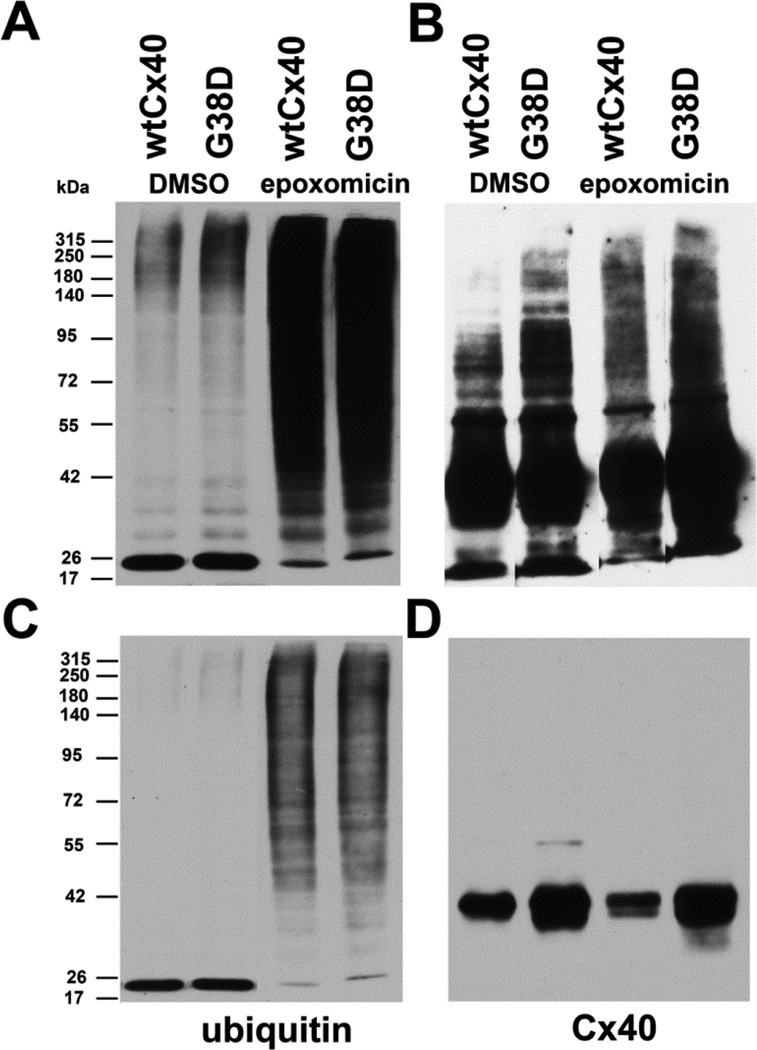
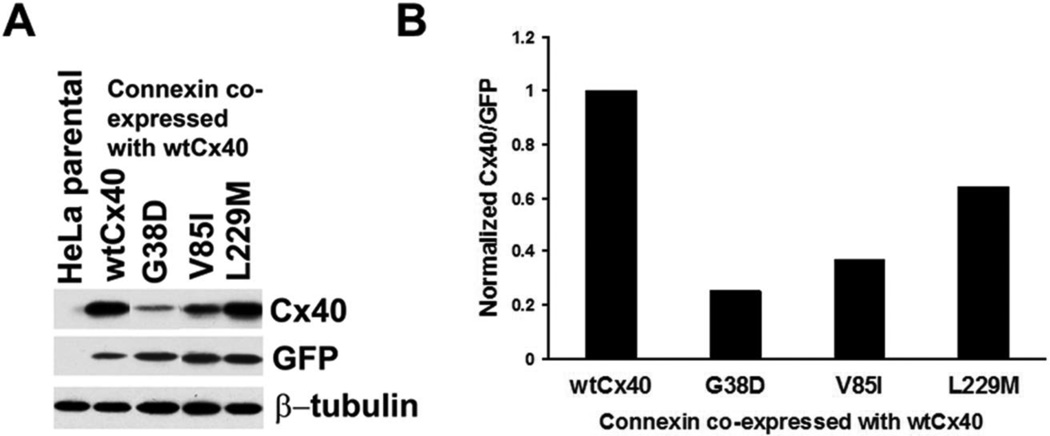
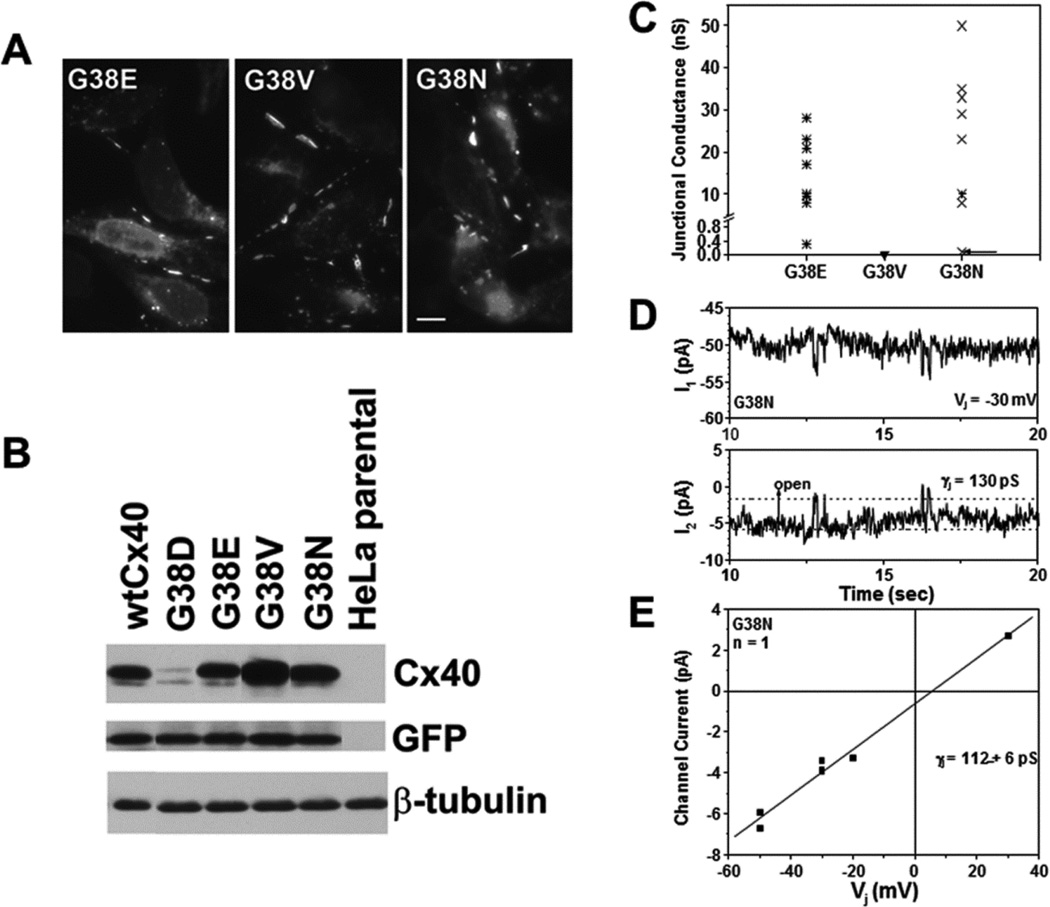
Several Cx40 mutants have been identified in patients with atrial fibrillation (AF). We have been working to identify physiological or cell biological abnormalities of several of these human mutants that might explain how they contribute to disease pathogenesis. Wild type (wt) Cx40 or four different mutants (P88S, G38D, V85I, and L229M) were expressed by the transfection of communication-deficient HeLa cells or HL-1 cardiomyocytes. Biophysical channel properties and the sub-cellular localization and protein levels of Cx40 were characterized. Wild type Cx40 and all mutants except P88S formed gap junction plaques and induced significant gap junctional conductances. The functional mutants showed only modest alterations of single channel conductances or gating by trans-junctional voltage as compared to wtCx40. However, immunoblotting indicated that the steady state levels of G38D, V85I, and L229M were reduced relative to wtCx40; most strikingly, G38D was only 20-31% of wild type levels. After the inhibition of protein synthesis with cycloheximide, G38D (and to a lesser extent the other mutants) disappeared much faster than wtCx40. Treatment with the proteasomal inhibitor, epoxomicin, greatly increased levels of G38D and restored the abundance of gap junctions and the extent of intercellular dye transfer. Thus, G38D, V85I, and L229M are functional mutants of Cx40 with small alterations of physiological properties, but accelerated degradation by the proteasome. These findings suggest a novel mechanism (protein instability) for the pathogenesis of AF due to a connexin mutation and a novel approach to therapy (protease inhibition).
Keywords: Atrial fibrillation; Connexin40; Gap junctions; Ion channels; Proteasome; Protein degradation.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Human Connexin40 Mutations Slow Conduction and Increase Propensity for Atrial Fibrillation.Heart Lung Circ. 2018 Jan;27(1):114-121. doi: 10.1016/j.hlc.2017.02.010. Epub 2017 Mar 21. Heart Lung Circ. 2018. PMID: 28457700
-
Altered conductance and permeability of Cx40 mutations associated with atrial fibrillation.J Gen Physiol. 2015 Nov;146(5):387-98. doi: 10.1085/jgp.201511475. J Gen Physiol. 2015. PMID: 26503720 Free PMC article.
-
Atrial fibrillation-associated connexin40 mutants make hemichannels and synergistically form gap junction channels with novel properties.FEBS Lett. 2014 Apr 17;588(8):1458-64. doi: 10.1016/j.febslet.2014.01.010. Epub 2014 Jan 20. FEBS Lett. 2014. PMID: 24457199 Free PMC article.
-
Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms.FEBS Lett. 2014 Apr 17;588(8):1238-43. doi: 10.1016/j.febslet.2014.02.064. Epub 2014 Mar 19. FEBS Lett. 2014. PMID: 24656738 Review.
-
Improving cardiac gap junction communication as a new antiarrhythmic mechanism: the action of antiarrhythmic peptides.Naunyn Schmiedebergs Arch Pharmacol. 2010 Mar;381(3):221-34. doi: 10.1007/s00210-009-0473-1. Epub 2009 Nov 27. Naunyn Schmiedebergs Arch Pharmacol. 2010. PMID: 19943035 Review.
Cited by
-
Connexin40 abnormalities and atrial fibrillation in the human heart.J Mol Cell Cardiol. 2014 Nov;76:159-68. doi: 10.1016/j.yjmcc.2014.08.021. Epub 2014 Sep 6. J Mol Cell Cardiol. 2014. PMID: 25200600 Free PMC article.
-
The effects of polyunsaturated fatty acids and antioxidant vitamins on atrial oxidative stress, nitrotyrosine residues, and connexins following extracorporeal circulation in patients undergoing cardiac surgery.Mol Cell Biochem. 2017 Sep;433(1-2):27-40. doi: 10.1007/s11010-017-3013-1. Epub 2017 Mar 23. Mol Cell Biochem. 2017. PMID: 28337705 Clinical Trial.
-
Helicobacter pylori regulates stomach diseases by activating cell pathways and DNA methylation of host cells.Front Cell Dev Biol. 2023 May 4;11:1187638. doi: 10.3389/fcell.2023.1187638. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 37215092 Free PMC article. Review.
-
Regulation of cardiac gap junctions by protein phosphatases.J Mol Cell Cardiol. 2017 Jun;107:52-57. doi: 10.1016/j.yjmcc.2017.05.002. Epub 2017 May 3. J Mol Cell Cardiol. 2017. PMID: 28478048 Free PMC article. Review.
-
Regulation of gap junction intercellular communication by connexin ubiquitination: physiological and pathophysiological implications.Cell Mol Life Sci. 2020 Feb;77(4):573-591. doi: 10.1007/s00018-019-03285-0. Epub 2019 Sep 9. Cell Mol Life Sci. 2020. PMID: 31501970 Free PMC article. Review.
References
-
- Allessie MA, Boyden PA, Camm AJ, Kleber AG, Lab MJ, Legato MJ, et al. Pathophysiology and prevention of atrial fibrillation. Circulation. 2001;103:769–777. - PubMed
-
- Iwasaki YK, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology: implications for management. Circulation. 2011;124:2264–2274. - PubMed
-
- Spach MS, Starmer CF. Altering the topology of gap junctions a major therapeutic target for atrial fibrillation. Cardiovasc Res. 1995;30:337–344. - PubMed
-
- Saffitz JE, Kanter HL, Green KG, Tolley TK, Beyer EC. Tissue-specific determinants of anisotropic conduction velocity in canine atrial and ventricular myocardium. Circ Res. 1994;74:1065–1070. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
