

Redox activation of DUSP4 by N-acetylcysteine protects endothelial cells from Cd²⁺-induced apoptosis
- PMID: 24973647
- PMCID: PMC4146716
- DOI: 10.1016/j.freeradbiomed.2014.06.016
Redox activation of DUSP4 by N-acetylcysteine protects endothelial cells from Cd²⁺-induced apoptosis
Abstract
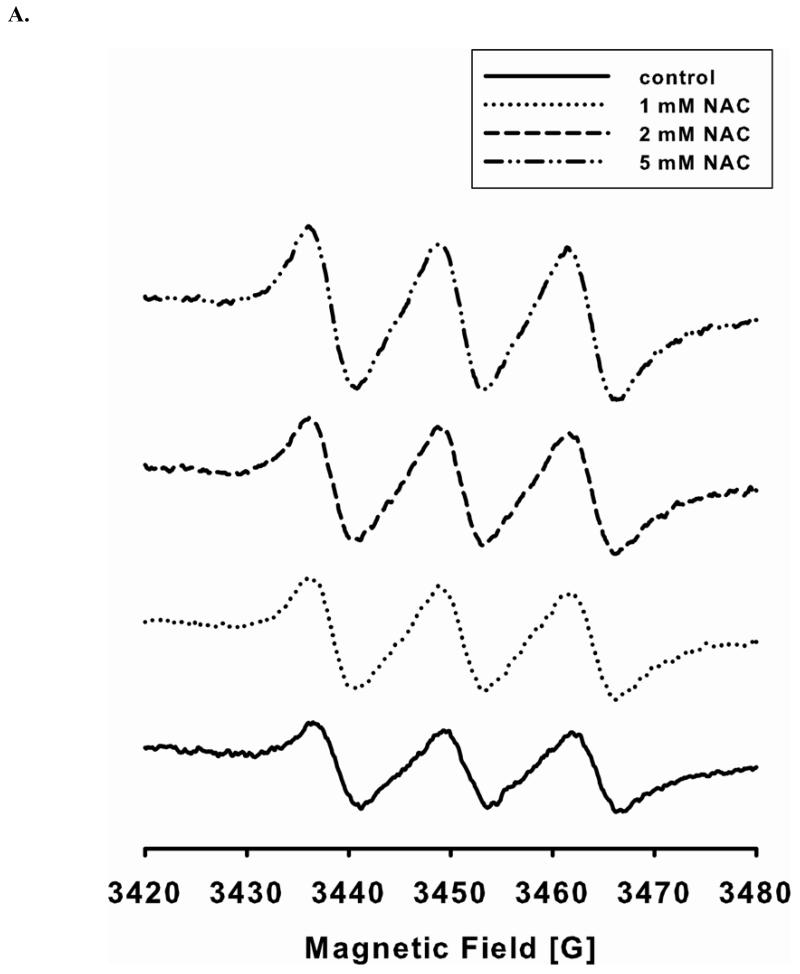
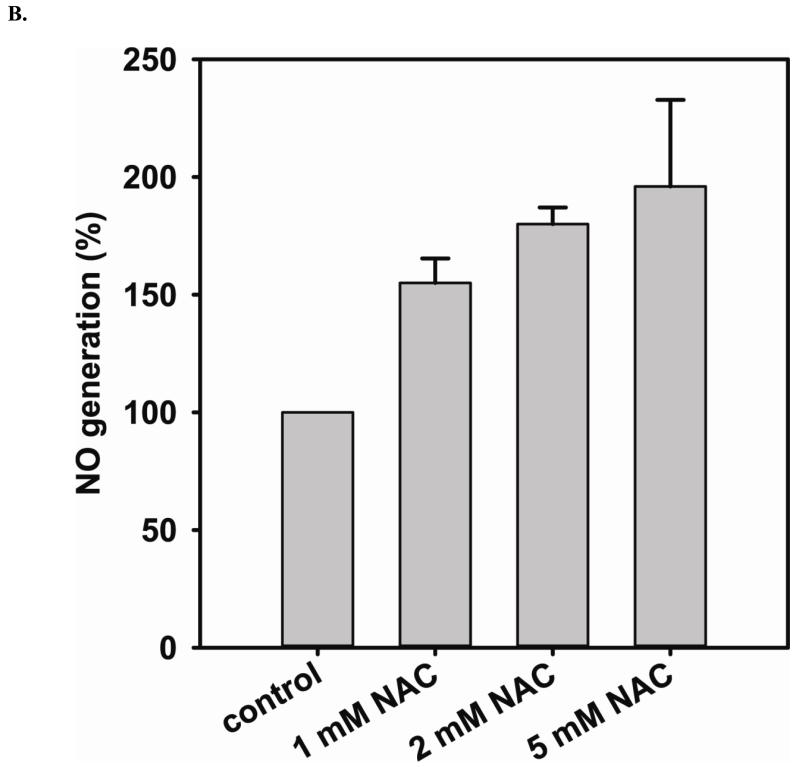
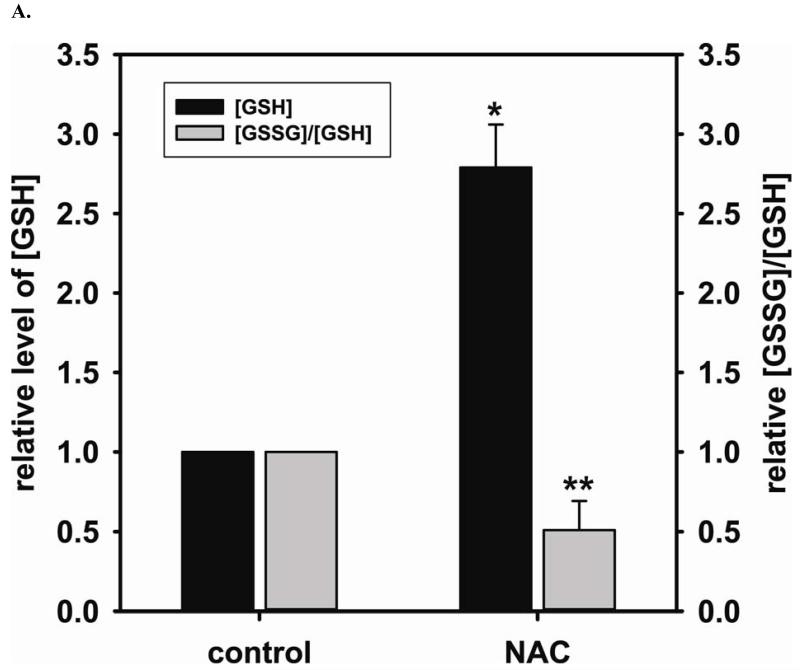
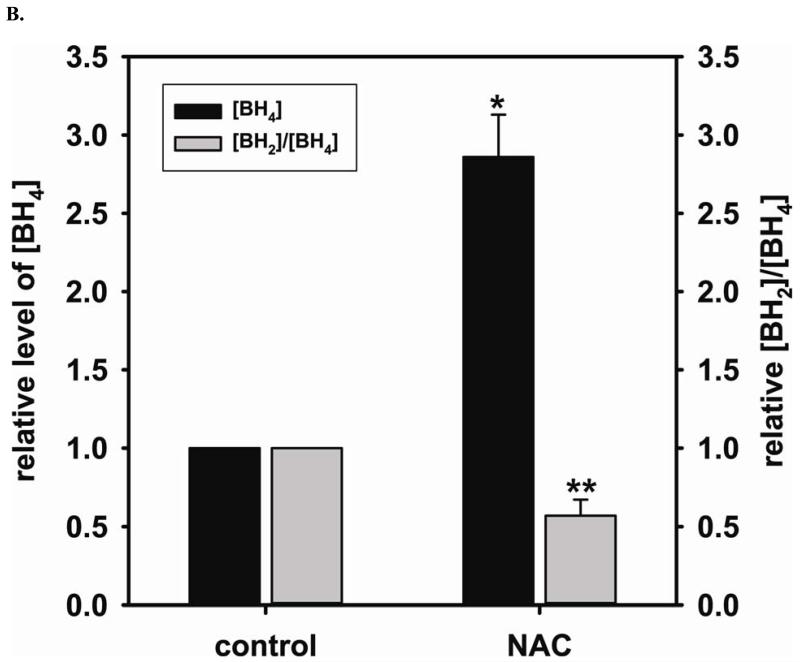
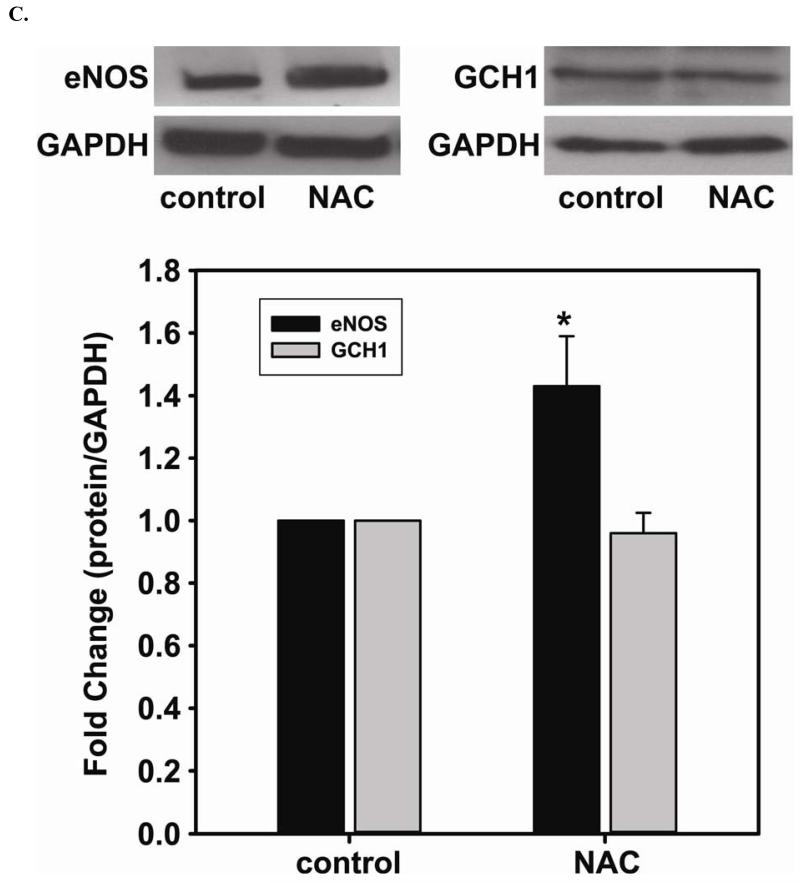
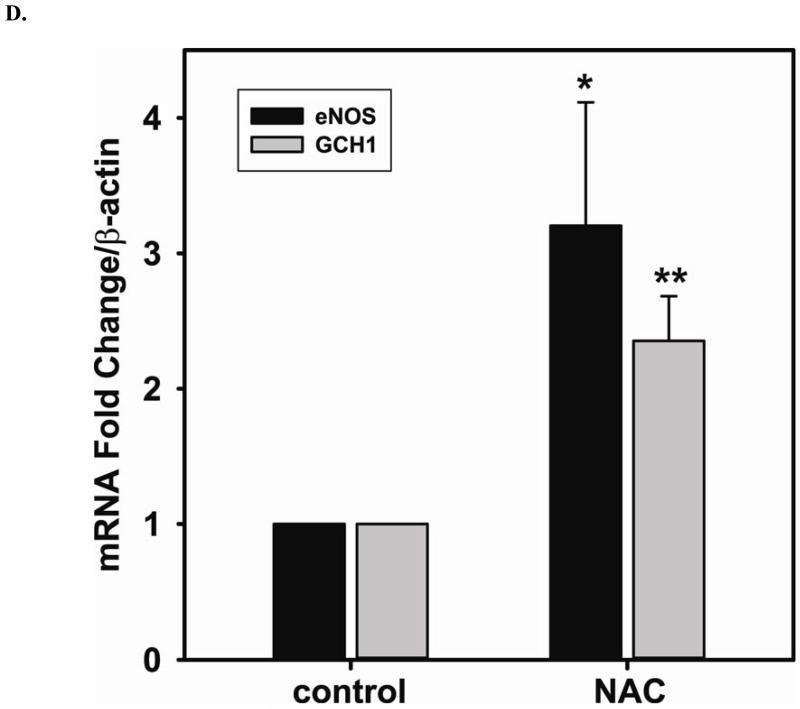
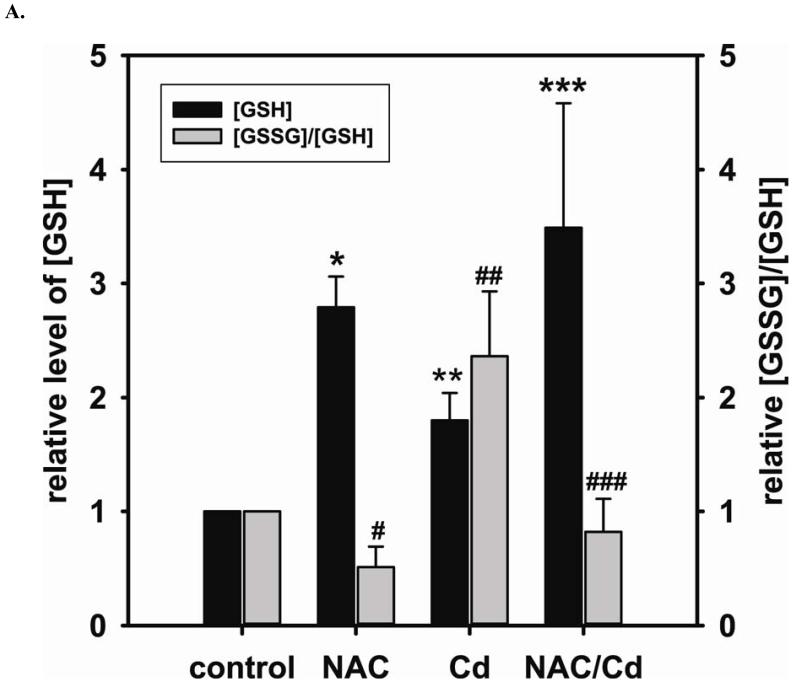
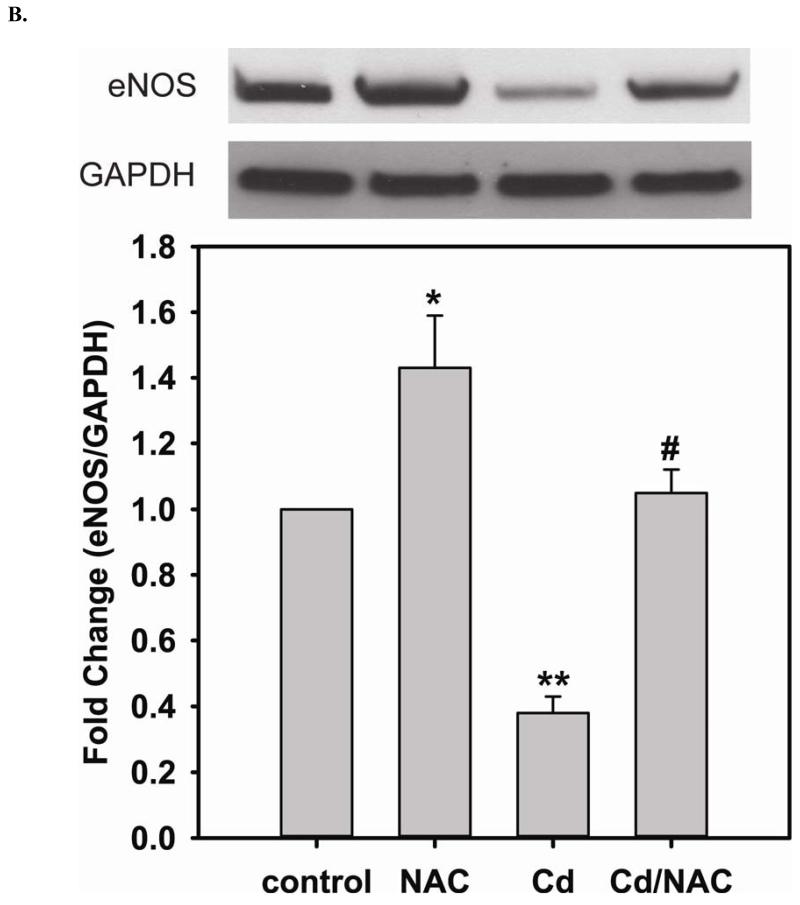
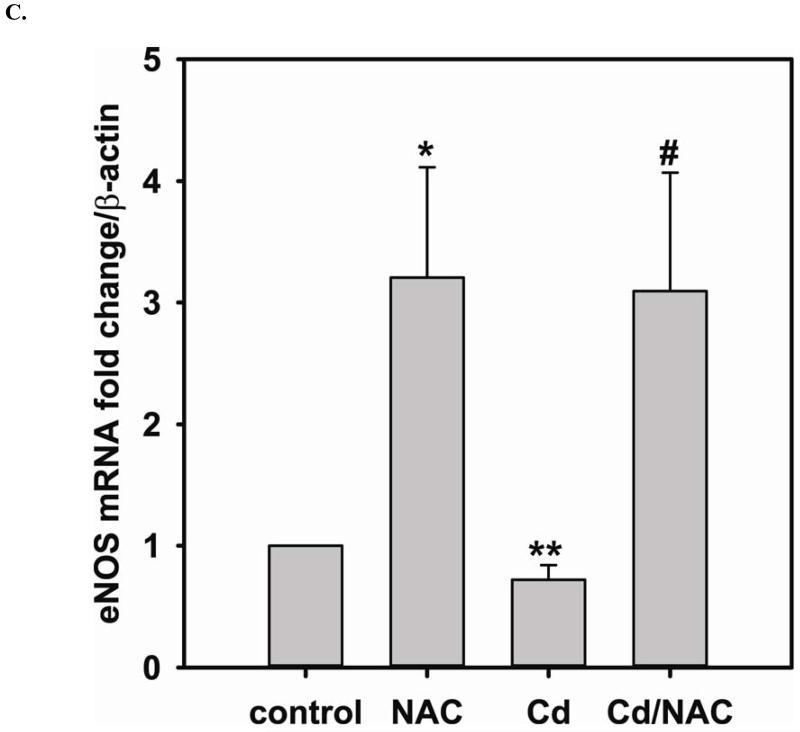
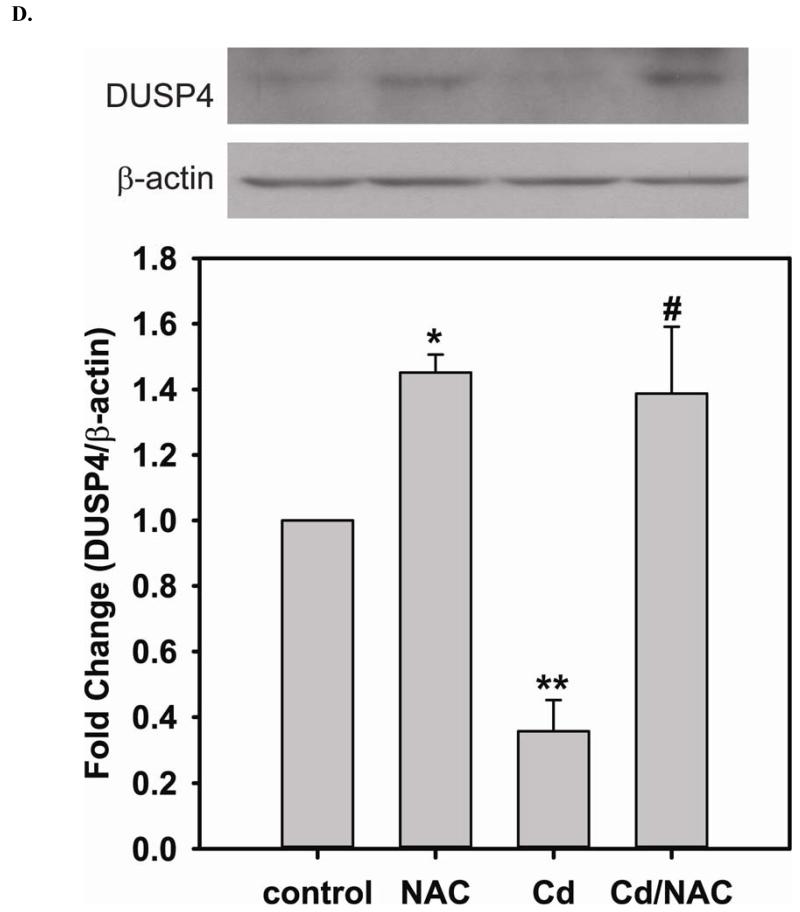
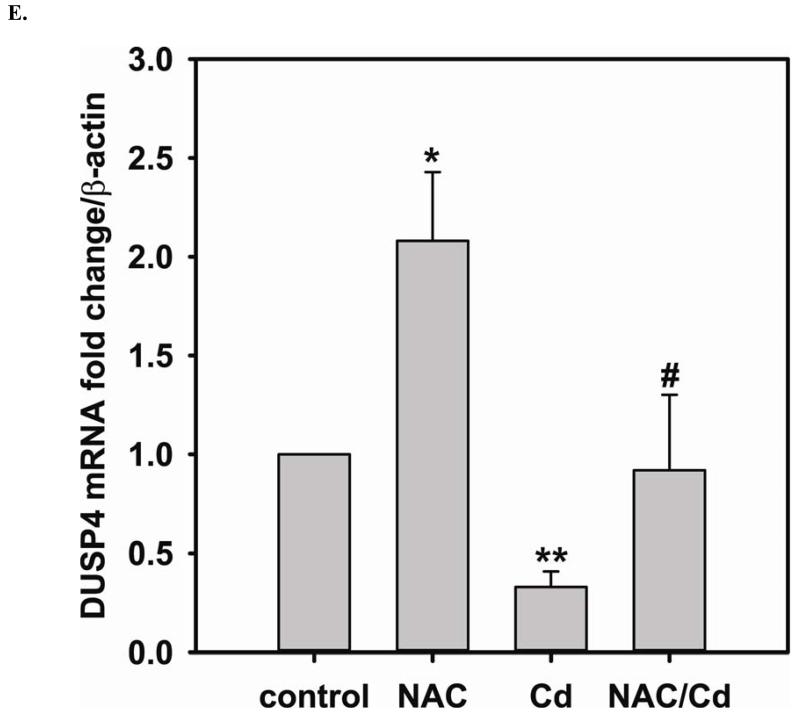
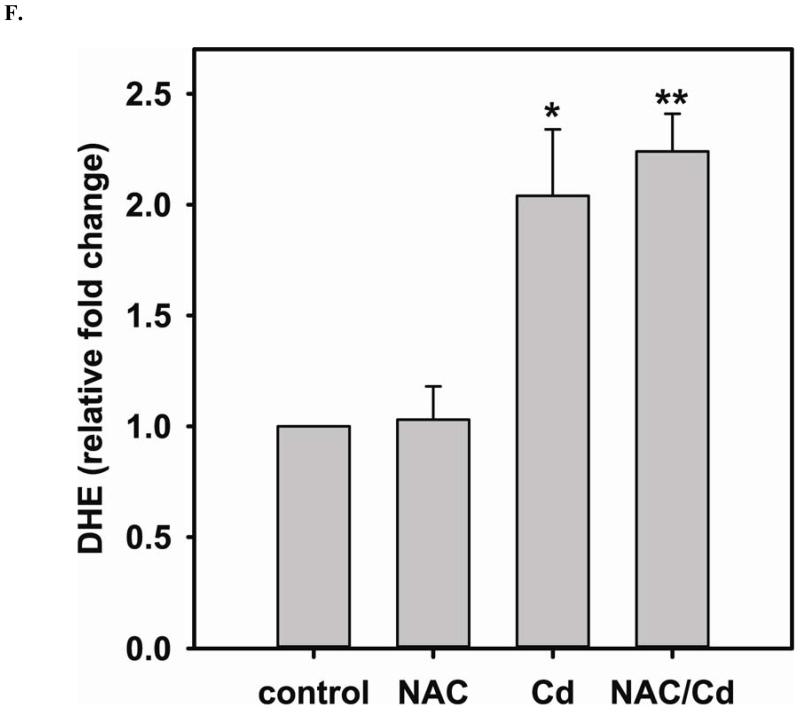
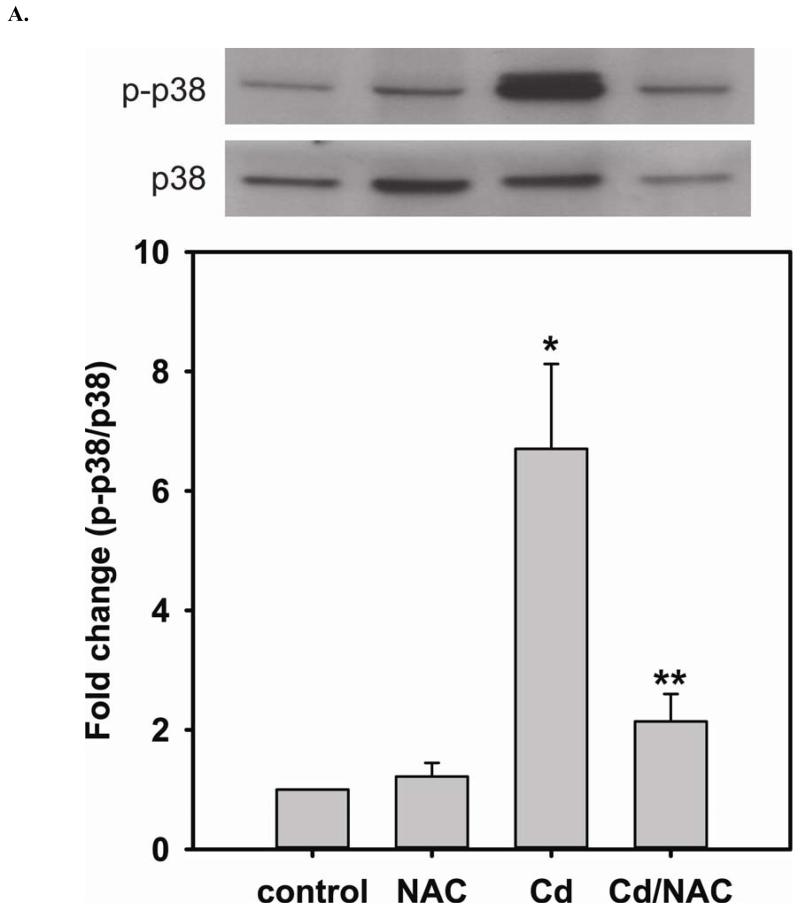
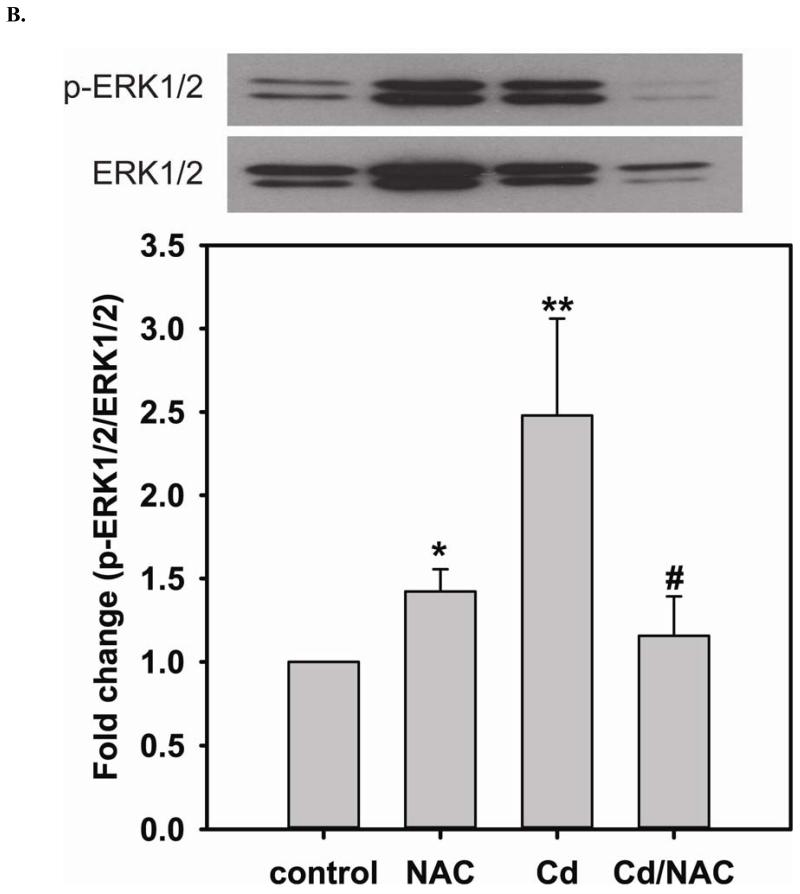
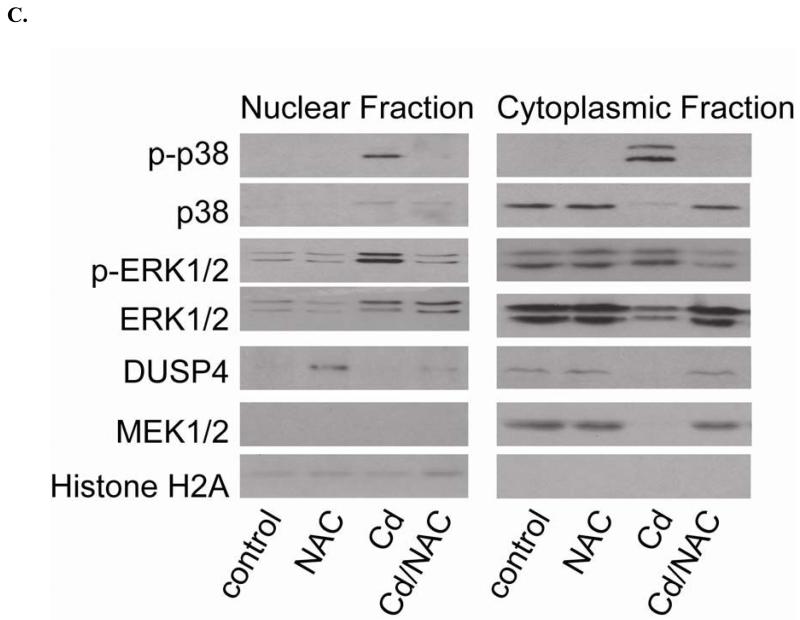
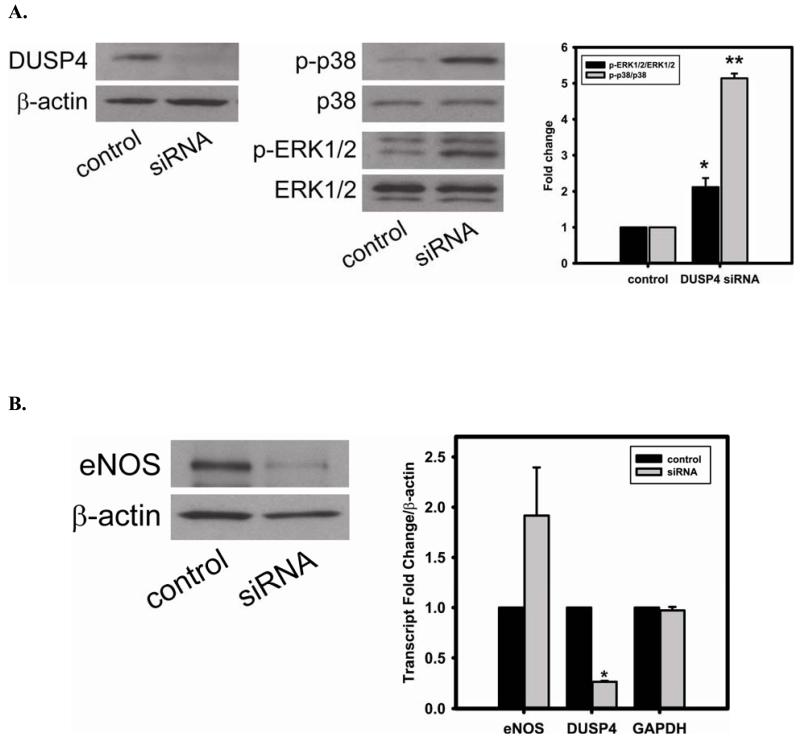
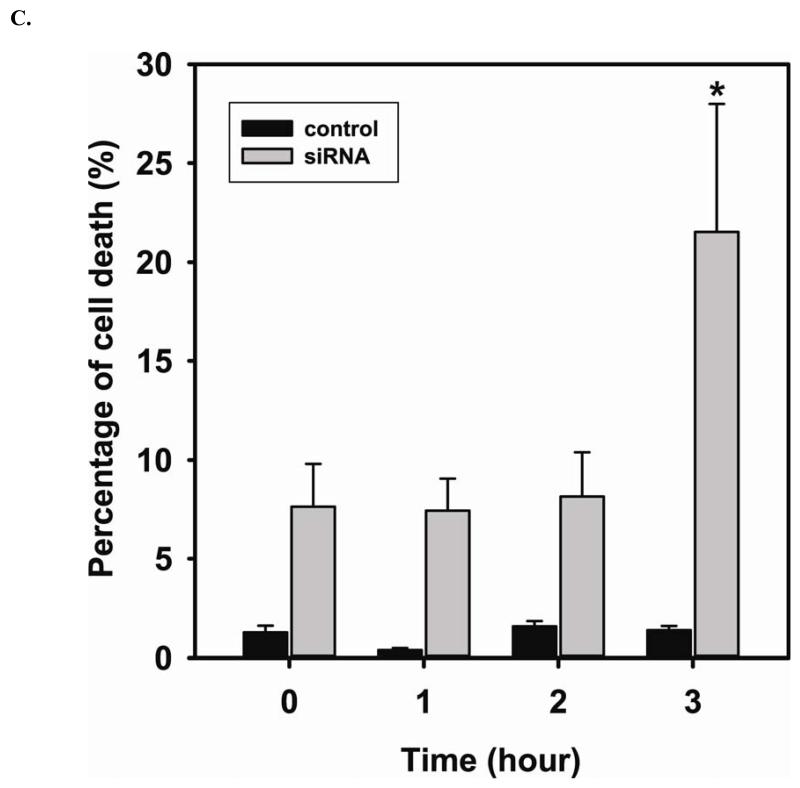
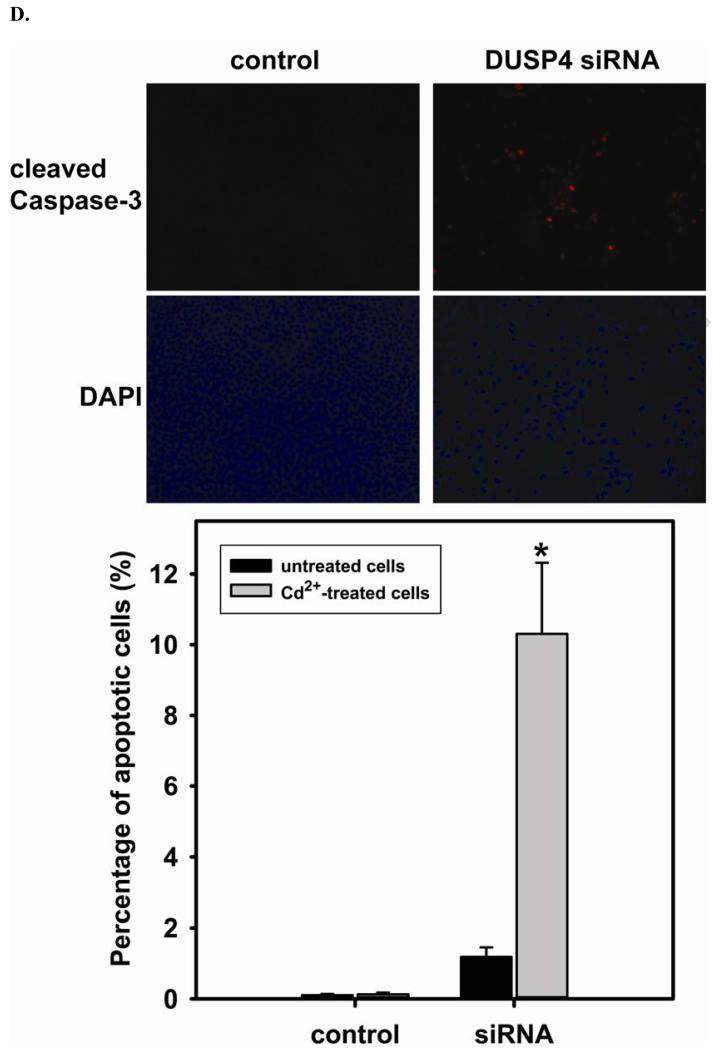
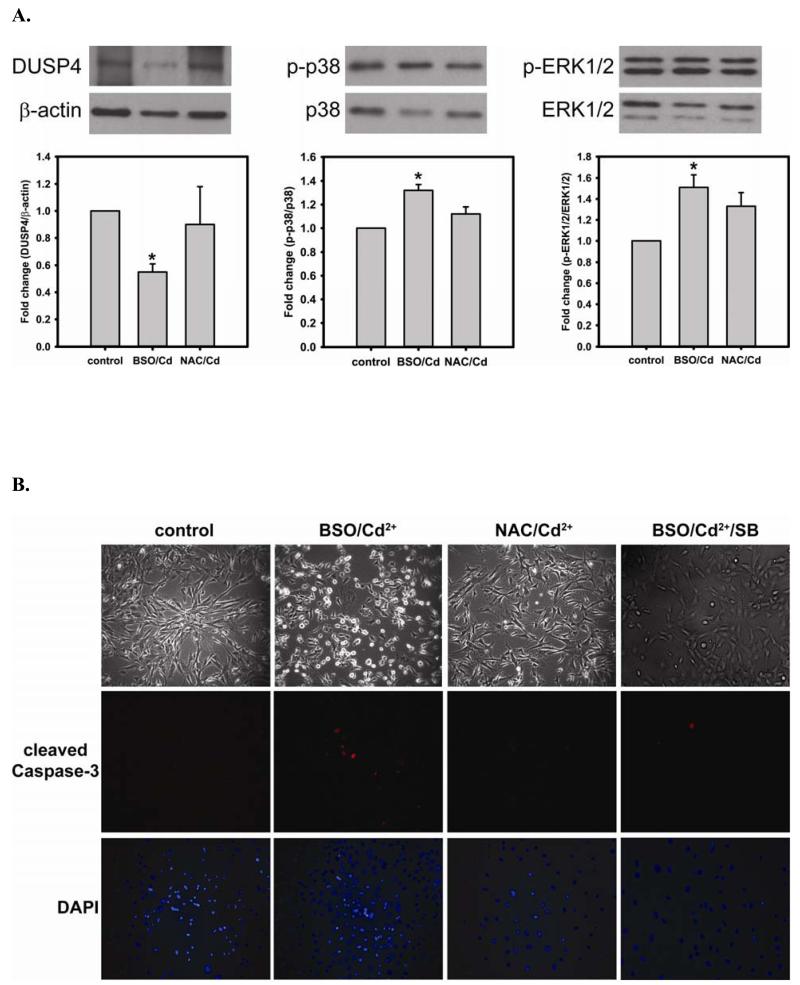
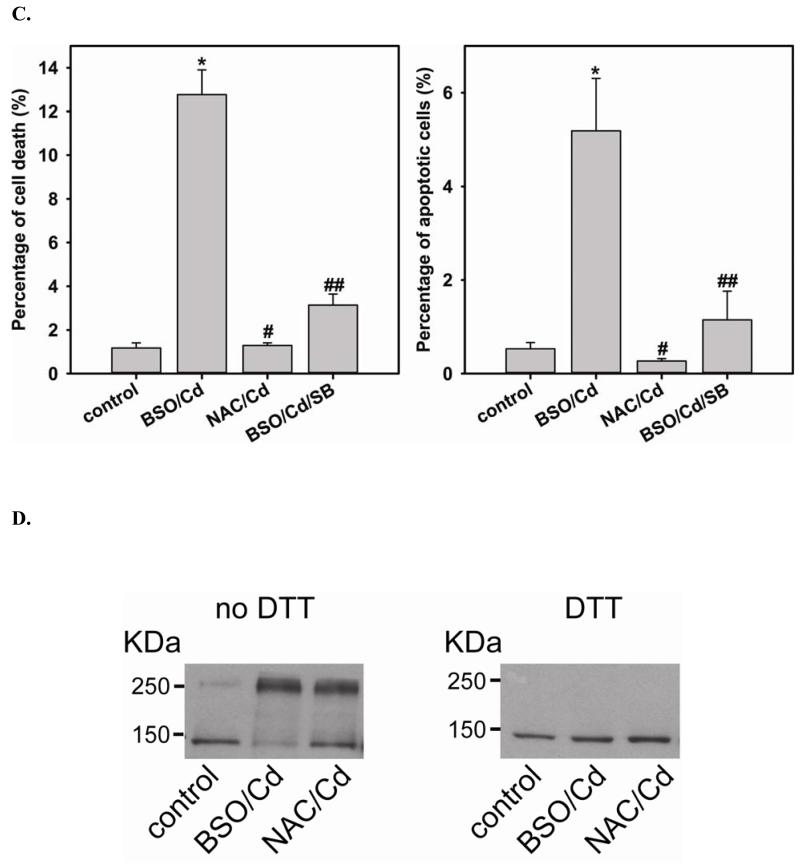
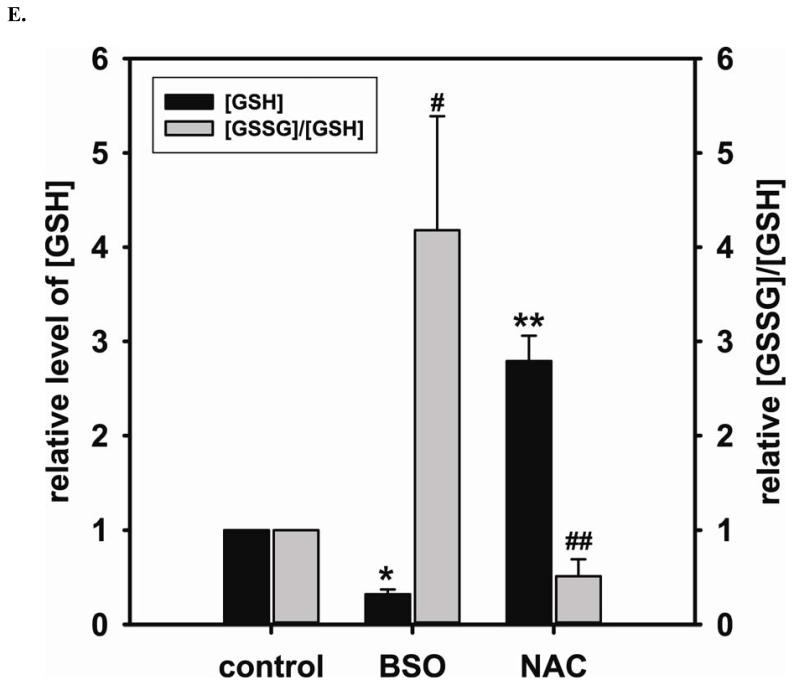
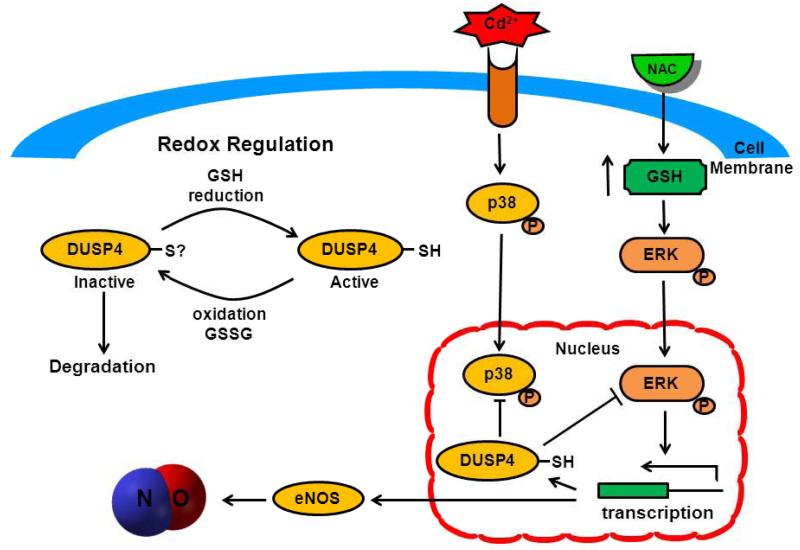
Redox imbalance is a primary cause of endothelial dysfunction (ED). Under oxidant stress, many critical proteins regulating endothelial function undergo oxidative modifications that lead to ED. Cellular levels of glutathione (GSH), the primary reducing source in cells, can significantly regulate cell function via reversible protein thiol modification. N-acetylcysteine (NAC), a precursor for GSH biosynthesis, is beneficial for many vascular diseases; however, the detailed mechanism of these benefits is still not clear. From HPLC analysis, NAC significantly increases both cellular GSH and tetrahydrobiopterin levels. Immunoblotting of endothelial NO synthase (eNOS) and DUSP4, a dual-specificity phosphatase with a cysteine as its active residue, revealed that both enzymes are upregulated by NAC. EPR spin trapping further demonstrated that NAC enhances NO generation from cells. Long-term exposure to Cd(2+) contributes to DUSP4 degradation and the uncontrolled activation of p38 and ERK1/2, leading to apoptosis. Treatment with NAC prevents DUSP4 degradation and protects cells against Cd(2+)-induced apoptosis. Moreover, the increased DUSP4 expression can redox-regulate the p38 and ERK1/2 pathways from hyperactivation, providing a survival mechanism against the toxicity of Cd(2+). DUSP4 gene knockdown further supports the hypothesis that DUSP4 is an antioxidant gene, critical in the modulation of eNOS expression, and thus protects against Cd(2+)-induced stress. Depletion of intracellular GSH by buthionine sulfoximine makes cells more susceptible to Cd(2+)-induced apoptosis. Pretreatment with NAC prevents p38 overactivation and thus protects the endothelium from this oxidative stress. Therefore, the identification of DUSP4 activation by NAC provides a novel target for future drug design.
Keywords: DUSP4; Endothelial dysfunction; Free radicals; MAP kinases; N-acetylcysteine; Nitric oxide synthase; Phosphatase; Reactive oxygen species; Redox signaling.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Archiv : European journal of physiology. 2010;459:32–939. - PubMed
-
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
