

Prospects for building large timetrees using molecular data with incomplete gene coverage among species
- PMID: 24974376
- PMCID: PMC4137717
- DOI: 10.1093/molbev/msu200
Prospects for building large timetrees using molecular data with incomplete gene coverage among species
Abstract
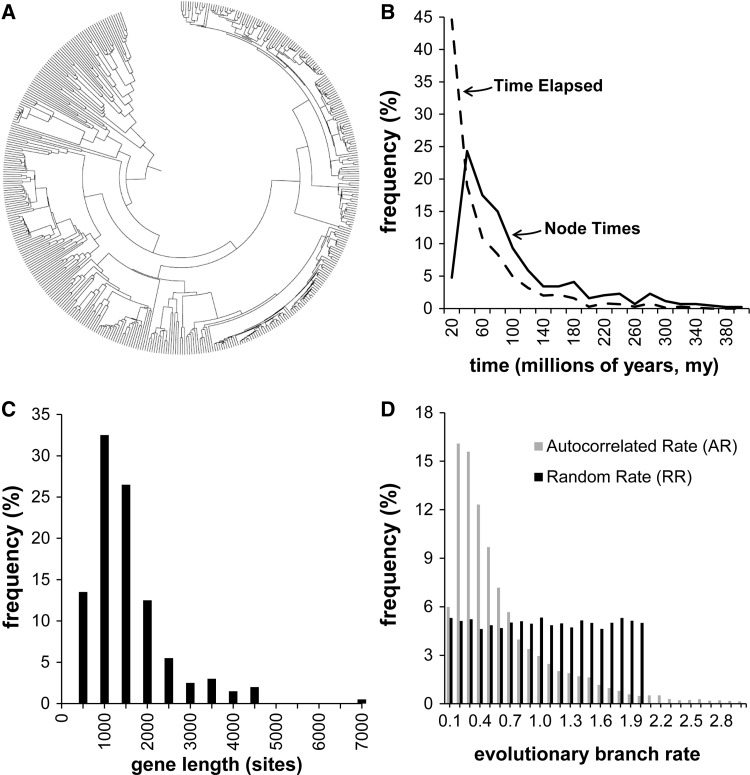
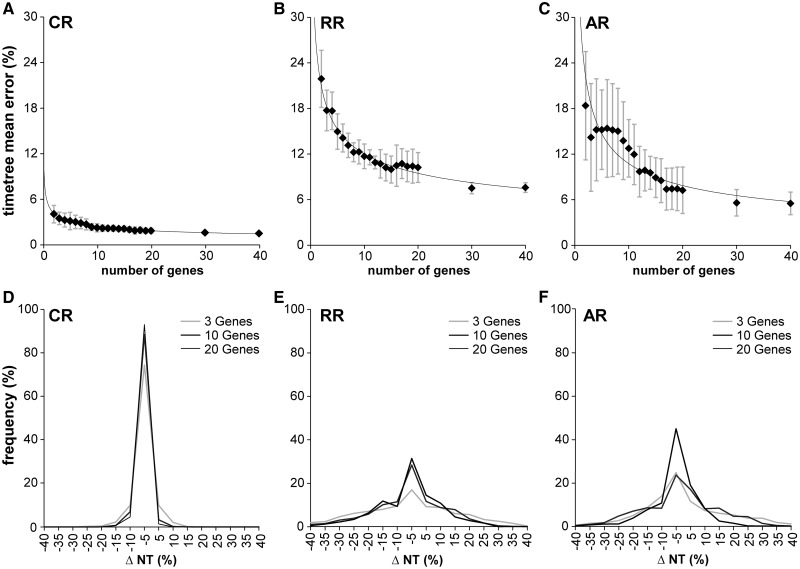
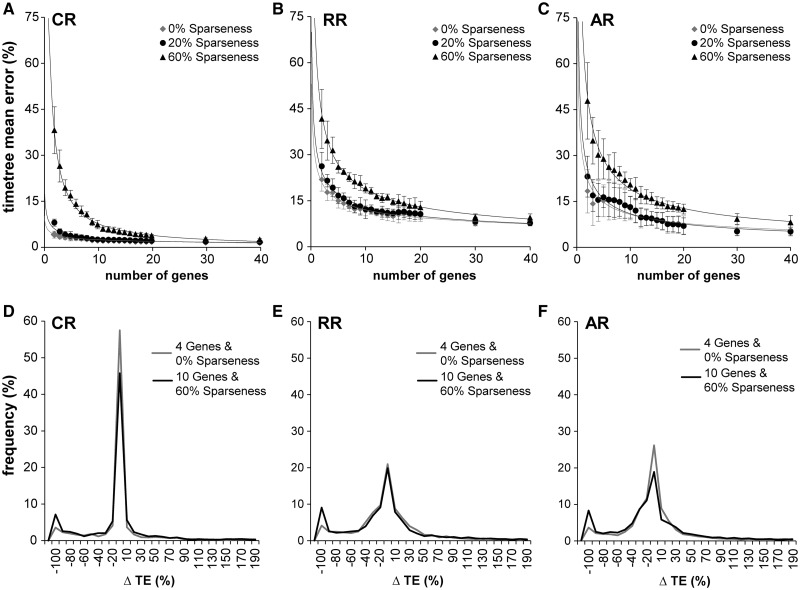
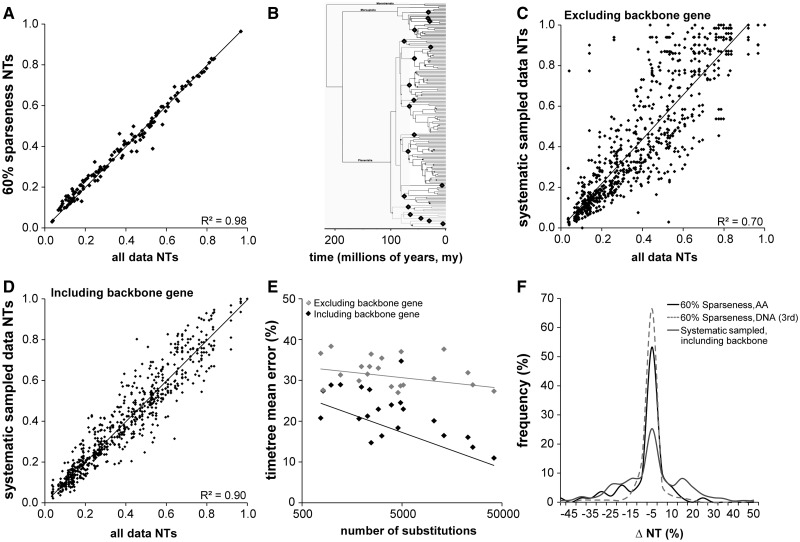
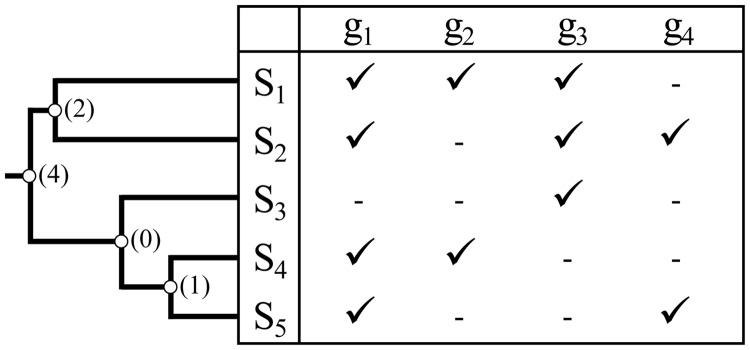
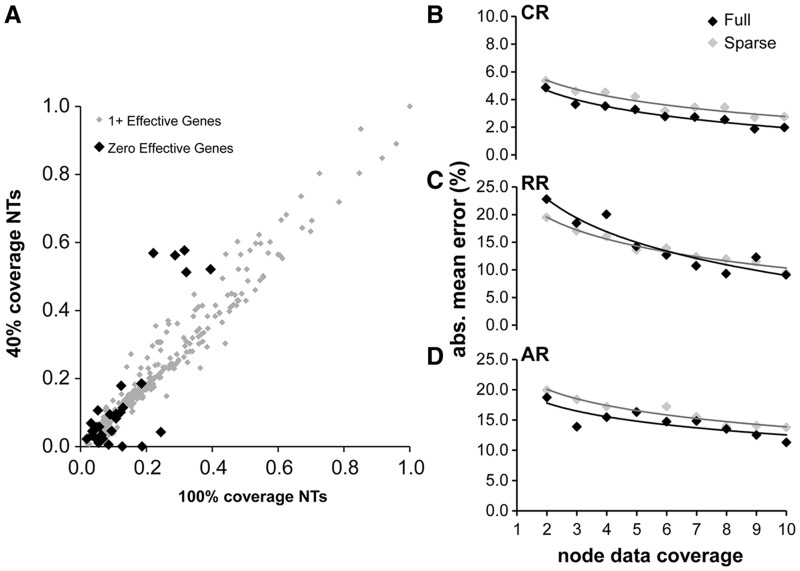
Scientists are assembling sequence data sets from increasing numbers of species and genes to build comprehensive timetrees. However, data are often unavailable for some species and gene combinations, and the proportion of missing data is often large for data sets containing many genes and species. Surprisingly, there has not been a systematic analysis of the effect of the degree of sparseness of the species-gene matrix on the accuracy of divergence time estimates. Here, we present results from computer simulations and empirical data analyses to quantify the impact of missing gene data on divergence time estimation in large phylogenies. We found that estimates of divergence times were robust even when sequences from a majority of genes for most of the species were absent. From the analysis of such extremely sparse data sets, we found that the most egregious errors occurred for nodes in the tree that had no common genes for any pair of species in the immediate descendant clades of the node in question. These problematic nodes can be easily detected prior to computational analyses based only on the input sequence alignment and the tree topology. We conclude that it is best to use larger alignments, because adding both genes and species to the alignment augments the number of genes available for estimating divergence events deep in the tree and improves their time estimates.
Keywords: divergence time; incomplete data; timetree.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Cracraft J, Donoghue MJ. Assembling the tree of life. New York: Oxford University Press; 2004.
-
- Douzery EJP, Delsuc F, Philippe H. Les datations moléculaires à l’heure de la génomique. Med Sci. 2006;22:374–380. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
