

The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival
- PMID: 24978191
- PMCID: PMC4076272
- DOI: 10.1371/journal.pone.0100516
The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival
Abstract
Background: Co-inheritance of α-thalassemia was reported to be associated with a delayed age of disease onset among Cameroonian Sickle Cell Anemia (SCA) patients. The present study aimed to explore the correlation between α-thalassemia, hematological indices, and clinical events in these patients.
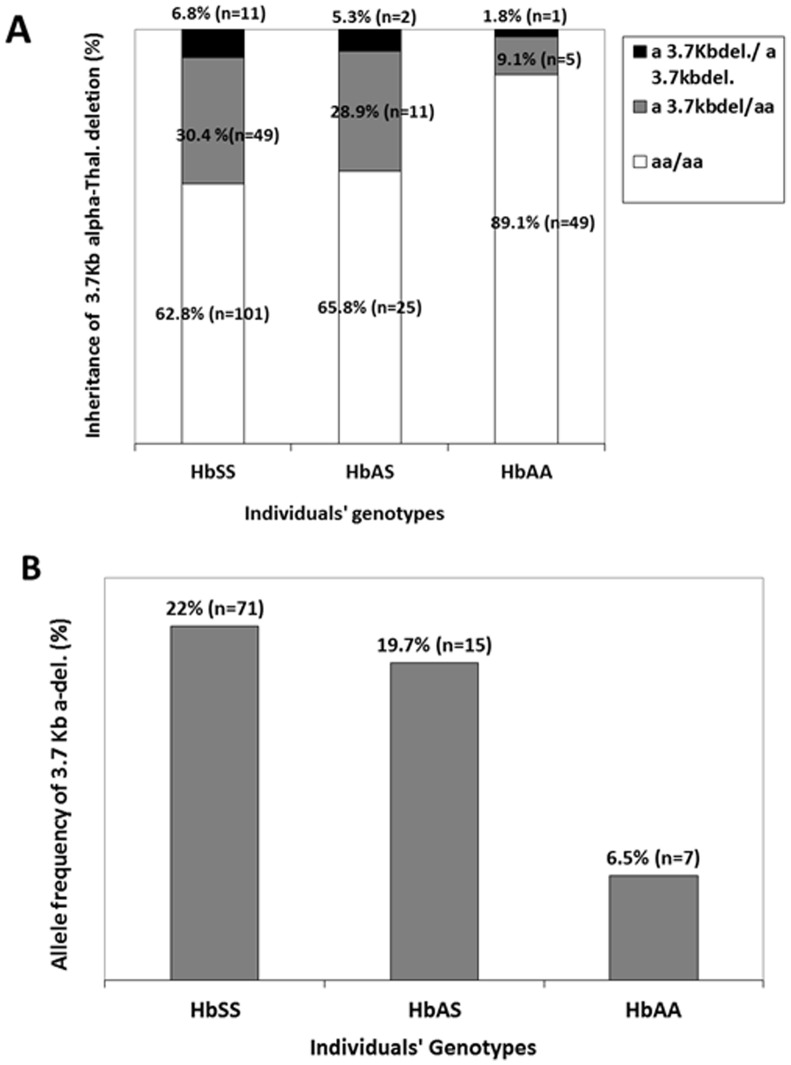
Methods and findings: We studied 161 Cameroonian SCA patients and 103 controls (59.1% HbAA) with median ages of 17.5 and 23 years. RFLP-PCR was used to confirm SCA genotype and to describe haplotypes in the HBB-like genes cluster. Multiplex Gap-PCR was performed to investigate the 3.7 kb α-globin gene deletions. SNaPshot PCR, capillary electrophoresis and cycle sequencing were used for the genotyping of 10 SNPs in BCL11A, HMIP1/2, OR51B5/6 and HBG loci, known to influence HbF levels. Generalised linear regression models adjusted for age, sex and SNPs genotypes was used to investigate effects of α-thalassemia on clinical and hematological indices. The median rate of vaso-occlusive painful crisis and hospitalisations was two and one per year, respectively. Stroke was reported in eight cases (7.4%). Benin haplotype was the most prevalent (66.3%; n = 208 chromosomes). Among patients, 37.3% (n = 60) had at least one 3.7 kb deletion, compared to 10.9% (n = 6) among HbAA controls (p<0.001). Among patients, the median RBC count increased with the number of 3.7 kb deletions [2.6, 3.0 and 3.4 million/dl, with no, one and two deletions (p = 0.01)]. The median MCV decreased with the number of 3.7 kb deletion [86, 80, and 68fl, with no, one and two deletions (p<0.0001)], as well as median WBC counts [13.2, 10.5 and 9.8×109/L (p<0.0001. The co-inheritance of α-thalassemia was associated with lower consultations rate (p = 0.038).
Conclusion: The co-inheritance of α-thalassemia and SCA is associated with improved hematological indices, and lower consultations rate in this group of patients. This could possibly improve their survival and explain the higher proportion of α-thalassemia among patients than controls.
Conflict of interest statement
Figures
References
-
- Bartolucci P, Galactéros F (2012) Clinical management of adult sickle-cell disease. Current Opinion in Hematology 19: 149–55. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
