

Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis
- PMID: 24980542
- PMCID: PMC4139065
- DOI: 10.1007/s11914-014-0223-2
Hypophosphatemic rickets: revealing novel control points for phosphate homeostasis
Abstract
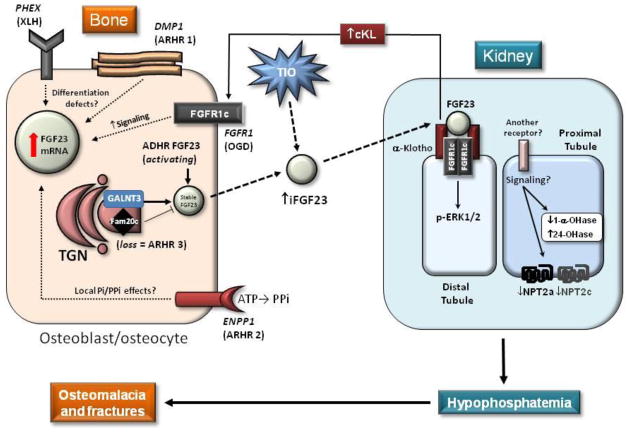
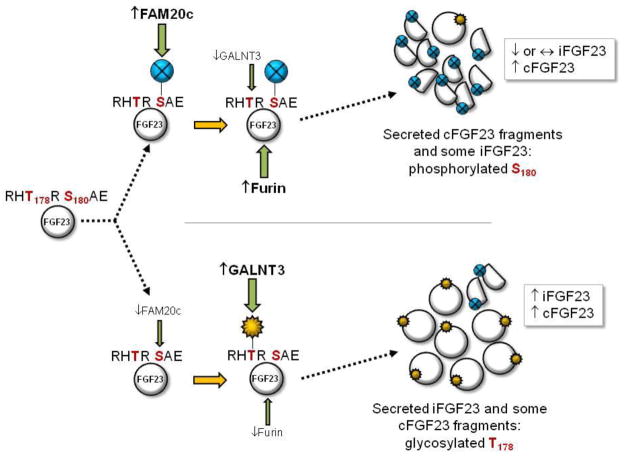
Rapid and somewhat surprising advances have recently been made toward understanding the molecular mechanisms causing heritable disorders of hypophosphatemia. The results of clinical, genetic, and translational studies have interwoven novel concepts underlying the endocrine control of phosphate metabolism, with far-reaching implications for treatment of both rare Mendelian diseases as well as common disorders of blood phosphate excess such as chronic kidney disease (CKD). In particular, diseases caused by changes in the expression and proteolytic control of the phosphaturic hormone fibroblast growth factor-23 (FGF23) have come to the forefront in terms of directing new models explaining mineral metabolism. These hypophosphatemic disorders as well as others resulting from independent defects in phosphate transport or metabolism will be reviewed herein, and implications for emerging therapeutic strategies based upon these new findings will be discussed.
Conflict of interest statement
KE White received royalties from Kyowa Hakko Kirin Co., Ltd. for licensing of the FGF23 gene and the anti-FGF23 monoclonal antibody trials.
JM Hum declares no conflicts of interest.
MJ Econs received royalties from Kyowa Hakko Kirin Co., Ltd. for licensing of the FGF23 gene and the anti-FGF23 monoclonal antibody trials and has also been a consultant to Kyowa Hakko Kirin for the anti-FGF23 monoclonal antibody trials.
Figures
References
-
- Tenenhouse HS, Econs MJ. Mendelian Hypophosphatemias. In: Valle D, editor. The Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies; New York: 2001. pp. 1–9.
-
- Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006;69(3):495–503. - PubMed
-
- Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, Quarles LD. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J Am Soc Nephrol. 2006;17(5):1305–15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
