

Ranking non-synonymous single nucleotide polymorphisms based on disease concepts
- PMID: 24980617
- PMCID: PMC4083756
- DOI: 10.1186/1479-7364-8-11
Ranking non-synonymous single nucleotide polymorphisms based on disease concepts
Abstract
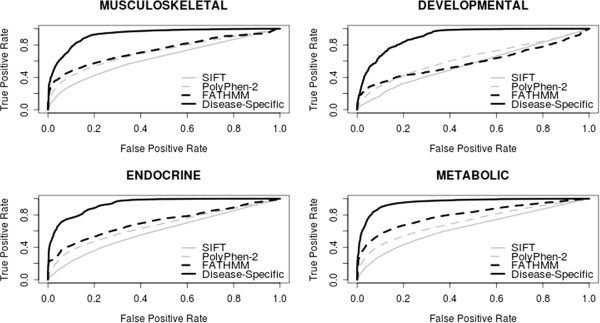
As the number of non-synonymous single nucleotide polymorphisms (nsSNPs) identified through whole-exome/whole-genome sequencing programs increases, researchers and clinicians are becoming increasingly reliant upon computational prediction algorithms designed to prioritize potential functional variants for further study. A large proportion of existing prediction algorithms are 'disease agnostic' but are nevertheless quite capable of predicting when a mutation is likely to be deleterious. However, most clinical and research applications of these algorithms relate to specific diseases and would therefore benefit from an approach that discriminates between functional variants specifically related to that disease from those which are not. In a whole-exome/whole-genome sequencing context, such an approach could substantially reduce the number of false positive candidate mutations. Here, we test this postulate by incorporating a disease-specific weighting scheme into the Functional Analysis through Hidden Markov Models (FATHMM) algorithm. When compared to traditional prediction algorithms, we observed an overall reduction in the number of false positives identified using a disease-specific approach to functional prediction across 17 distinct disease concepts/categories. Our results illustrate the potential benefits of making disease-specific predictions when prioritizing candidate variants in relation to specific diseases. A web-based implementation of our algorithm is available at http://fathmm.biocompute.org.uk.
Figures
References
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. - PubMed
-
- Thusberg J, Olatubosun A, Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat. 2011;32:358–368. - PubMed
-
- Sasidharan Nair P, Vihinen M. VariBench: a benchmark database for variations. Hum Mutat. 2013;34:42–49. - PubMed
-
- Kaminker JS, Zhang Y, Waugh A, Haverty PM, Peters B, Sebisanovic D, Stinson J, Forrest WF, Bazan JF, Seshagiri S, Zhang Z. Distinguishing cancer-associated missense mutations from common polymorphisms. Cancer Res. 2007;67:465–473. - PubMed
Publication types
MeSH terms
Grants and funding
- G1000427/1/MRC_/Medical Research Council/United Kingdom
- BB/G022771/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MC_UU_12013/8/MRC_/Medical Research Council/United Kingdom
- G1000427/MRC_/Medical Research Council/United Kingdom
- BB/G022771/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
