

SnowyOwl: accurate prediction of fungal genes by using RNA-Seq and homology information to select among ab initio models
- PMID: 24980894
- PMCID: PMC4084796
- DOI: 10.1186/1471-2105-15-229
SnowyOwl: accurate prediction of fungal genes by using RNA-Seq and homology information to select among ab initio models
Abstract
Background: Locating the protein-coding genes in novel genomes is essential to understanding and exploiting the genomic information but it is still difficult to accurately predict all the genes. The recent availability of detailed information about transcript structure from high-throughput sequencing of messenger RNA (RNA-Seq) delineates many expressed genes and promises increased accuracy in gene prediction. Computational gene predictors have been intensively developed for and tested in well-studied animal genomes. Hundreds of fungal genomes are now or will soon be sequenced. The differences of fungal genomes from animal genomes and the phylogenetic sparsity of well-studied fungi call for gene-prediction tools tailored to them.
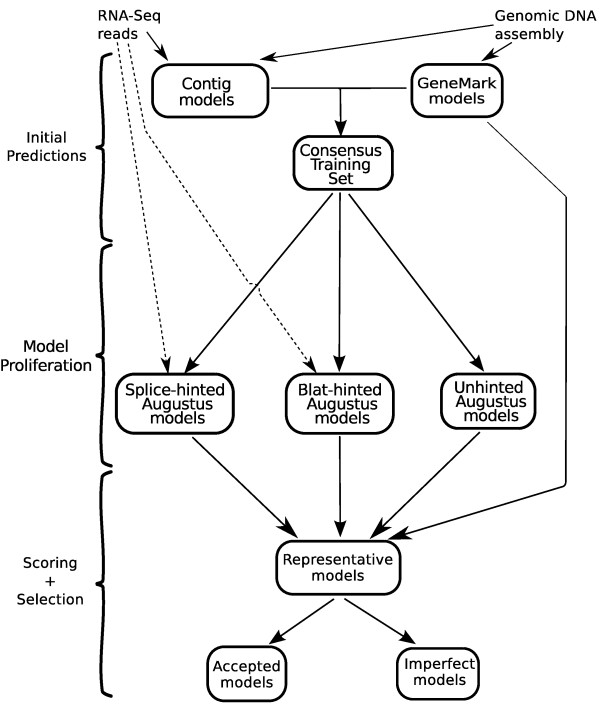
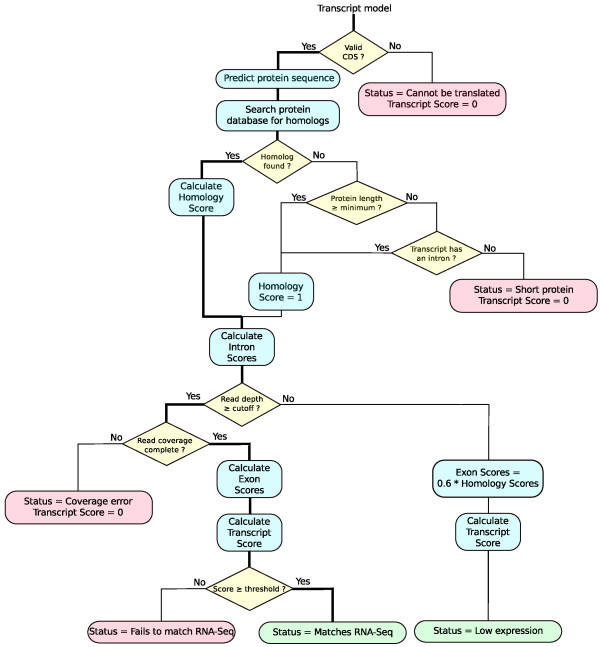
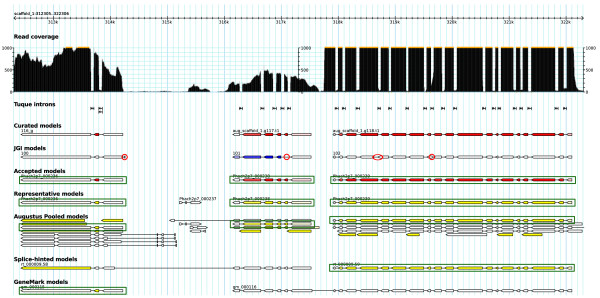
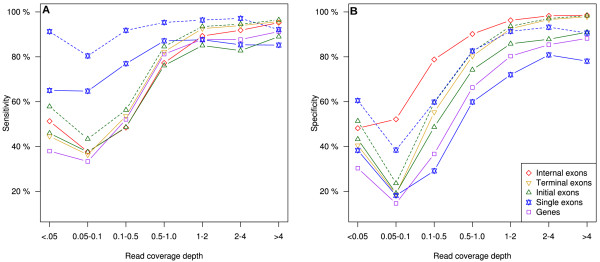
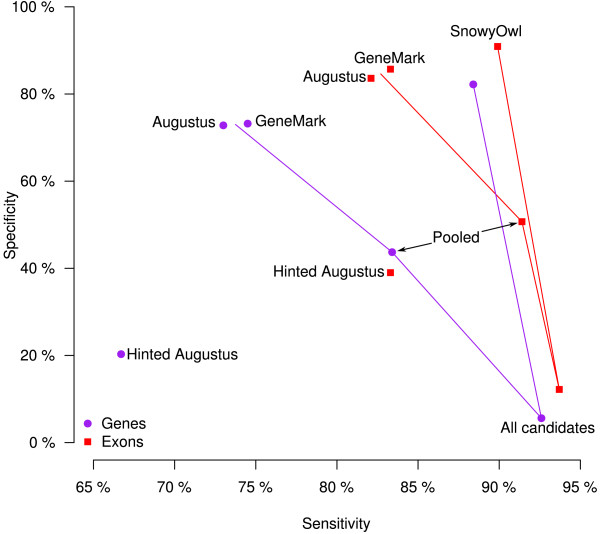
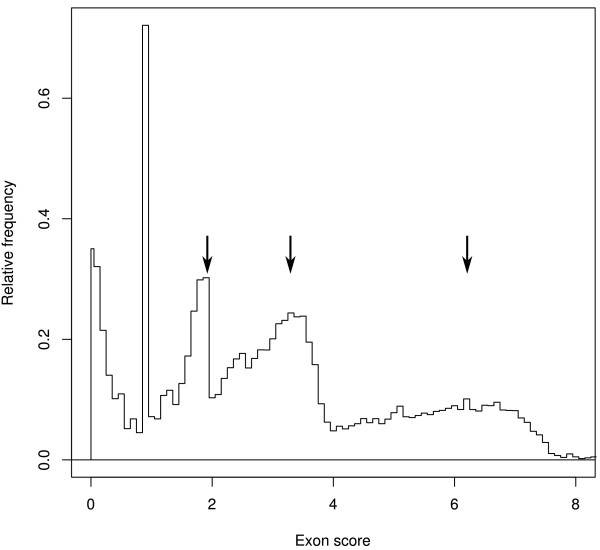
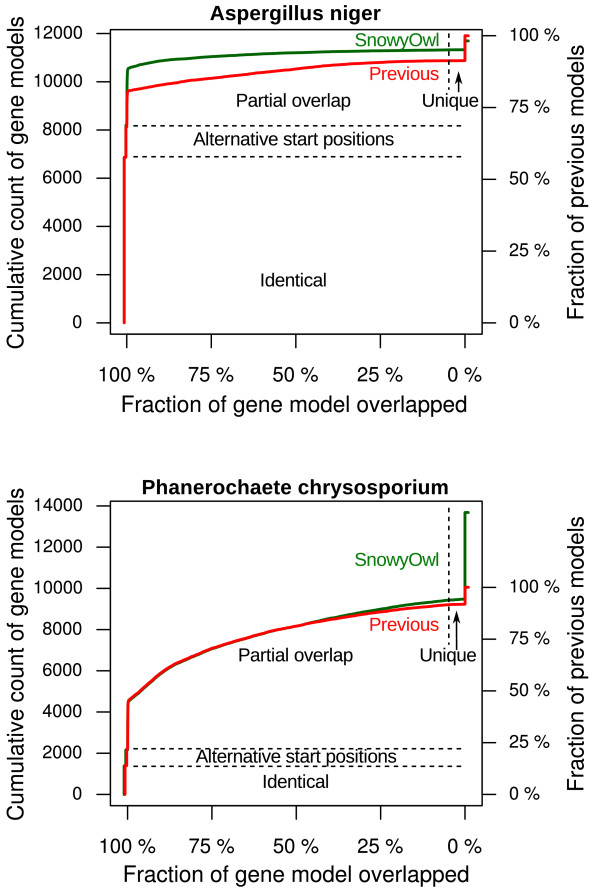
Results: SnowyOwl is a new gene prediction pipeline that uses RNA-Seq data to train and provide hints for the generation of Hidden Markov Model (HMM)-based gene predictions and to evaluate the resulting models. The pipeline has been developed and streamlined by comparing its predictions to manually curated gene models in three fungal genomes and validated against the high-quality gene annotation of Neurospora crassa; SnowyOwl predicted N. crassa genes with 83% sensitivity and 65% specificity. SnowyOwl gains sensitivity by repeatedly running the HMM gene predictor Augustus with varied input parameters and selectivity by choosing the models with best homology to known proteins and best agreement with the RNA-Seq data.
Conclusions: SnowyOwl efficiently uses RNA-Seq data to produce accurate gene models in both well-studied and novel fungal genomes. The source code for the SnowyOwl pipeline (in Python) and a web interface (in PHP) is freely available from http://sourceforge.net/projects/snowyowl/.
Figures
References
-
- Majoros WH. Methods for Computational Gene Prediction. New York: Cambridge University Press; 2007.
-
- Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–ii225. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
