

The route to pathologies in chronic inflammatory diseases characterized by T helper type 2 immune cells
- PMID: 24981014
- PMCID: PMC4233369
- DOI: 10.1111/cei.12409
The route to pathologies in chronic inflammatory diseases characterized by T helper type 2 immune cells
Abstract
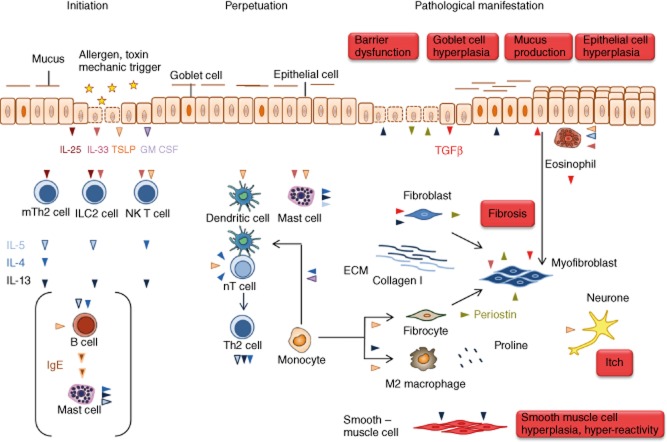
T helper type 2 (Th2)-characterized inflammatory responses are highly dynamic processes initiated by epithelial cell damage resulting in remodelling of the tissue architecture to prevent further harm caused by a dysfunctional epithelial barrier or migrating parasites. This process is a temporal and spatial response which requires communication between immobile cells such as epithelial, endothelial, fibroblast and muscle cells and the highly mobile cells of the innate and adaptive immunity. It is further characterized by a high cellular plasticity that enables the cells to adapt to a specific inflammatory milieu. Incipiently, this milieu is shaped by cytokines released from epithelial cells, which stimulate Th2, innate lymphoid and invariant natural killer (NK) T cells to secrete Th2 cytokines and to activate dendritic cells which results in the further differentiation of Th2 cells. This milieu promotes wound-healing processes which are beneficial in parasitic infections or toxin exposure but account for increasingly dysfunctional vital organs, such as the lung in the case of asthma and the colon in ulcerative colitis. A better understanding of the dynamics underlying relapses and remissions might lead ultimately to improved therapeutics for chronic inflammatory diseases adapted to individual needs and to different phases of the inflammation.
Keywords: Th1/Th2 cells; cytokine receptors; cytokines; dendritic cells; inflammation.
© 2014 British Society for Immunology.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
