

Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling
- PMID: 24982443
- PMCID: PMC4150064
- DOI: 10.1242/jcs.150458
Conformational remodeling of the fibronectin matrix selectively regulates VEGF signaling
Abstract
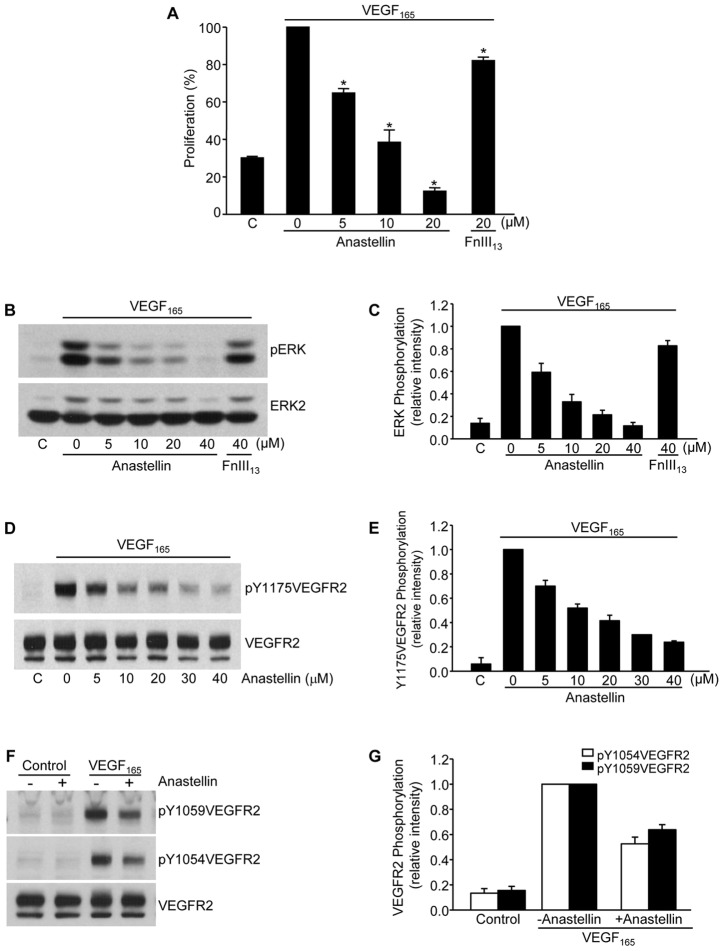
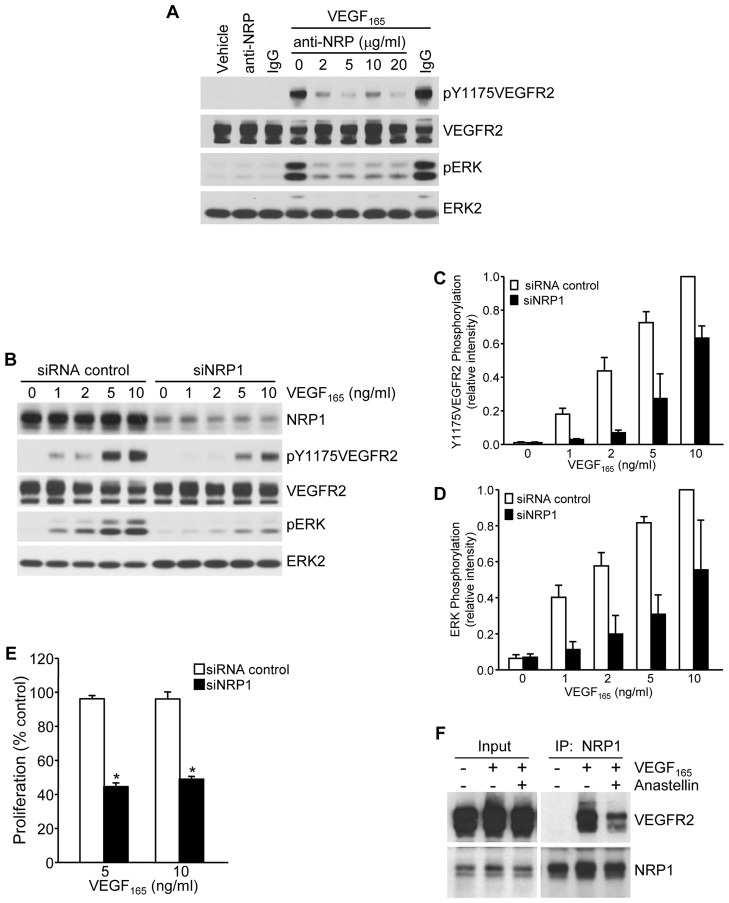
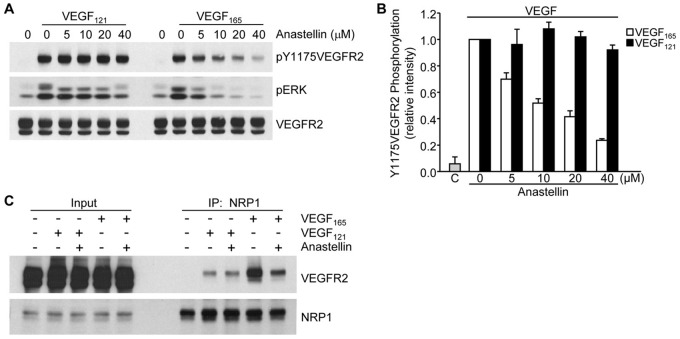
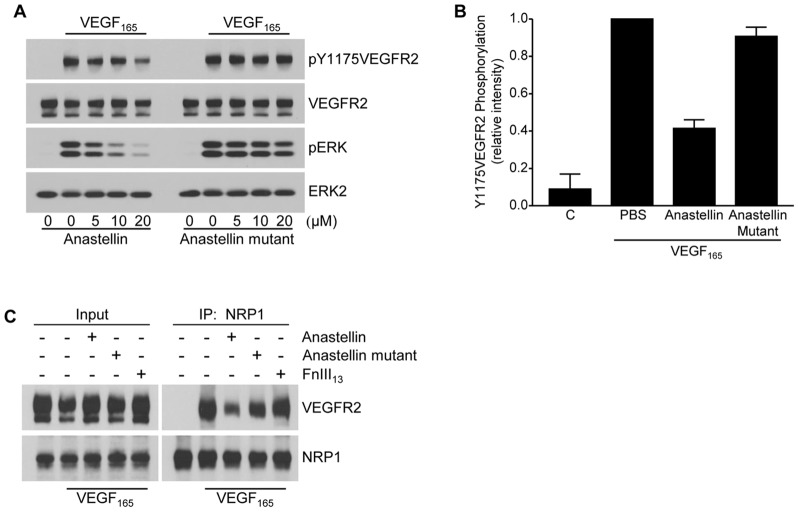
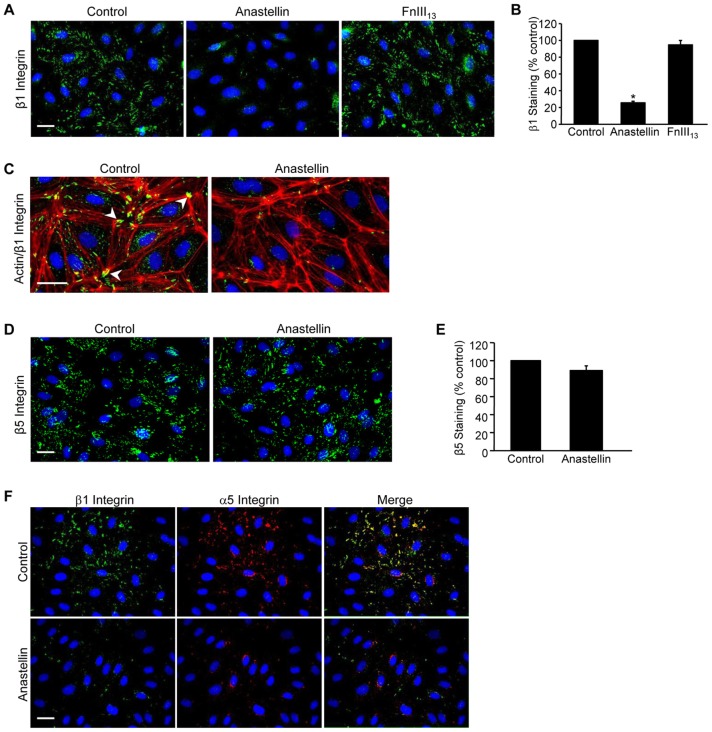
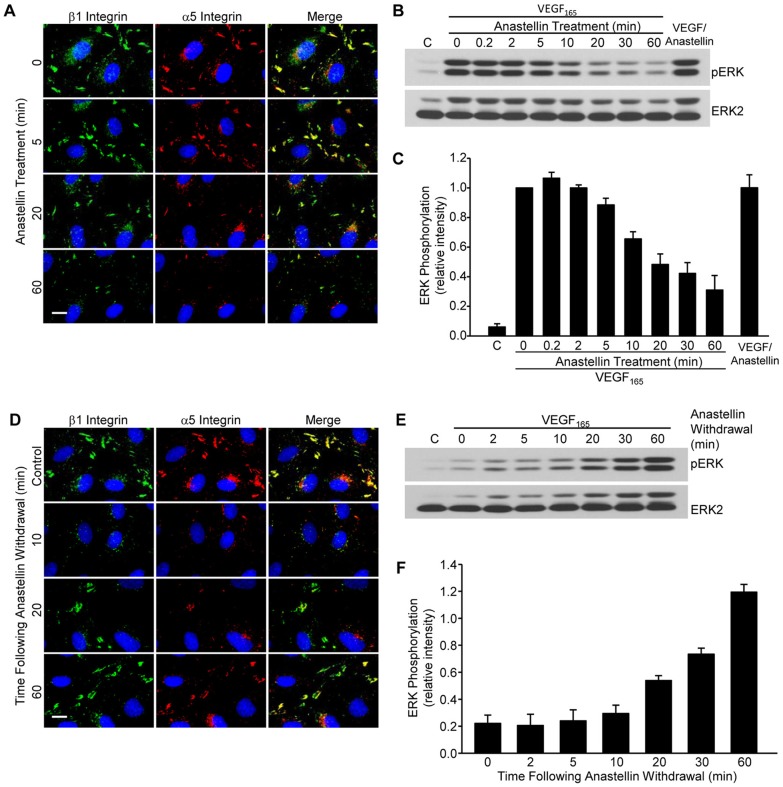
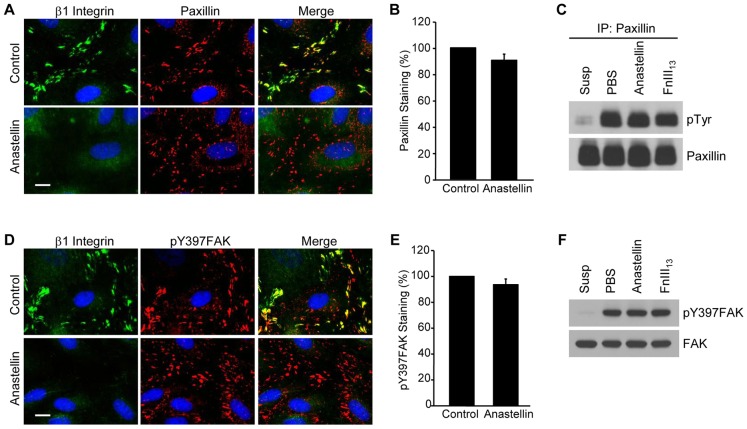
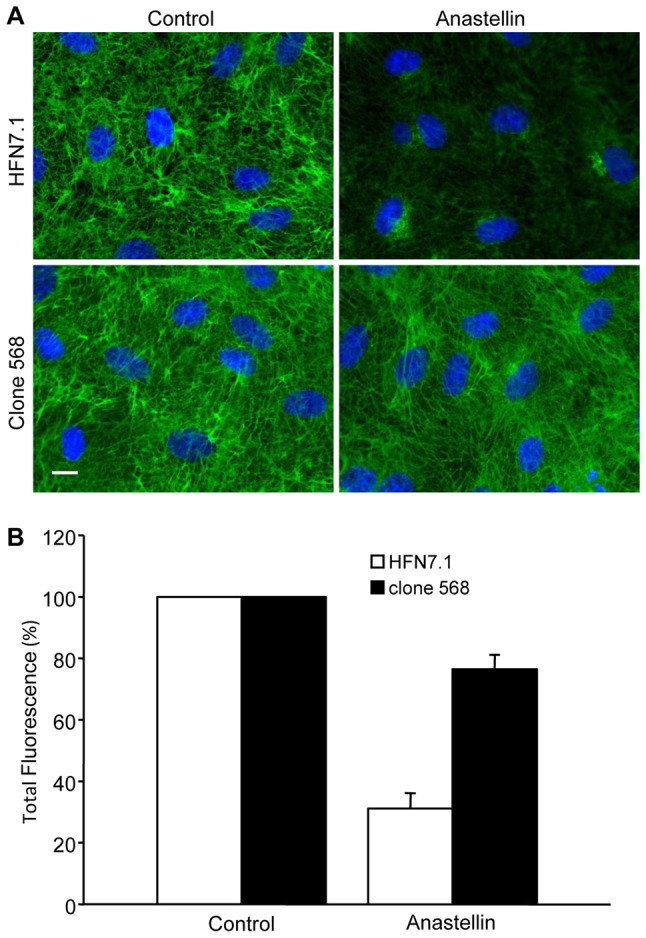
The fibronectin matrix plays a crucial role in the regulation of angiogenesis during development, tissue repair and pathogenesis. Previous work has identified a fibronectin-derived homophilic binding peptide, anastellin, as an effective inhibitor of angiogenesis; however, its mechanism of action is not well understood. In the present study, we demonstrate that anastellin selectively inhibits microvessel cell signaling in response to the VEGF165 isoform, but not VEGF121, by preventing the assembly of the complex containing the VEGF receptor and neuropilin-1. Anastellin treatment resulted in the inactivation of α5β1 integrins but was not accompanied by a change in either adhesion complexes or adhesion-based signaling. Integrin inactivation was associated with a masking of the fibronectin synergy site within the extracellular matrix (ECM), indicating that α5β1 inactivation resulted from a decrease in available ligand. These data demonstrate that anastellin influences the microvessel cell response to growth factors by controlling the repertoire of ligated integrins and point to anastellin as an effective regulator of fibronectin matrix organization. These studies further suggest that homophilic fibronectin binding peptides might have novel applications in the field of tissue regeneration as tools to regulate neovascularization.
Keywords: Anastellin; Angiogenesis; Extracellular matrix; Fibronectin; Integrin; Neuropilin; VEGF.
© 2014. Published by The Company of Biologists Ltd.
Figures
References
-
- Ambesi A., Klein R. M., Pumiglia K. M., McKeown-Longo P. J. (2005). Anastellin, a fragment of the first type III repeat of fibronectin, inhibits extracellular signal-regulated kinase and causes G(1) arrest in human microvessel endothelial cells. Cancer Res. 65, 148–156 - PubMed
-
- Aota S., Nomizu M., Yamada K. M. (1994). The short amino acid sequence Pro-His-Ser-Arg-Asn in human fibronectin enhances cell-adhesive function. J. Biol. Chem. 269, 24756–24761 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
