

HLA diversity in the 1000 genomes dataset
- PMID: 24988075
- PMCID: PMC4079705
- DOI: 10.1371/journal.pone.0097282
HLA diversity in the 1000 genomes dataset
Abstract
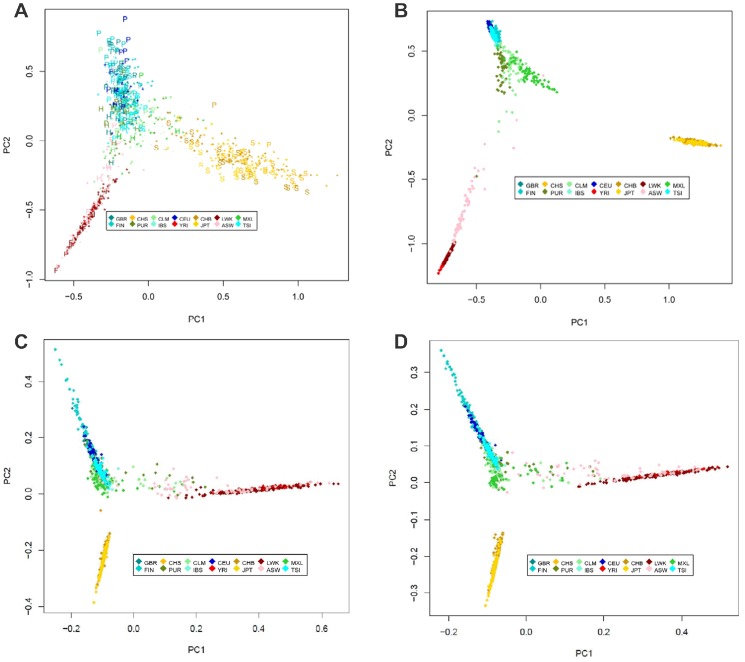
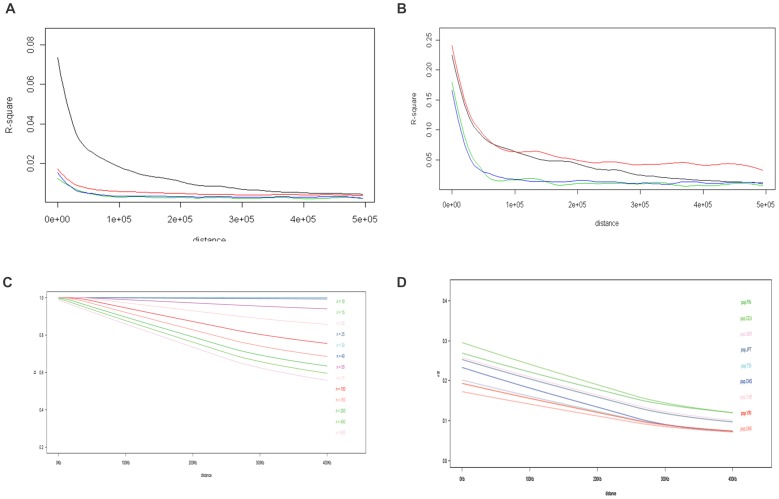
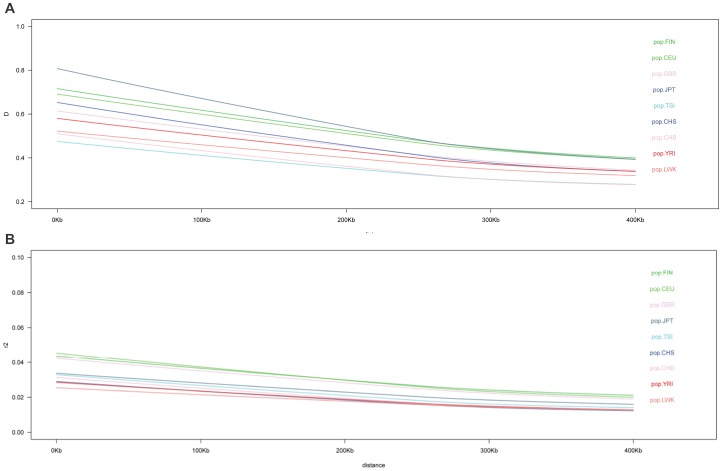
The 1000 Genomes Project aims to provide a deep characterization of human genome sequence variation by sequencing at a level that should allow the genome-wide detection of most variants with frequencies as low as 1%. However, in the major histocompatibility complex (MHC), only the top 10 most frequent haplotypes are in the 1% frequency range whereas thousands of haplotypes are present at lower frequencies. Given the limitation of both the coverage and the read length of the sequences generated by the 1000 Genomes Project, the highly variable positions that define HLA alleles may be difficult to identify. We used classical Sanger sequencing techniques to type the HLA-A, HLA-B, HLA-C, HLA-DRB1 and HLA-DQB1 genes in the available 1000 Genomes samples and combined the results with the 103,310 variants in the MHC region genotyped by the 1000 Genomes Project. Using pairwise identity-by-descent distances between individuals and principal component analysis, we established the relationship between ancestry and genetic diversity in the MHC region. As expected, both the MHC variants and the HLA phenotype can identify the major ancestry lineage, informed mainly by the most frequent HLA haplotypes. To some extent, regions of the genome with similar genetic or similar recombination rate have similar properties. An MHC-centric analysis underlines departures between the ancestral background of the MHC and the genome-wide picture. Our analysis of linkage disequilibrium (LD) decay in these samples suggests that overestimation of pairwise LD occurs due to a limited sampling of the MHC diversity. This collection of HLA-specific MHC variants, available on the dbMHC portal, is a valuable resource for future analyses of the role of MHC in population and disease studies.
Conflict of interest statement
Figures
References
-
- Horton R, Wilming L, Rand V, Lovering RC, Bruford EA, et al. (2004) Gene map of the extended human MHC. Nat Rev Genet 5: 889–899. - PubMed
-
- Susal C, Opelz G (2012) Impact of HLA Matching and HLA Antibodies in Organ Transplantation: A Collaborative Transplant Study View. Methods Mol Biol 882: 267–277. - PubMed
-
- van Rood JJ, Oudshoorn M (2008) Eleven million donors in Bone Marrow Donors Worldwide! Time for reassessment? Bone Marrow Transplant 41: 1–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
