

Consensus proposals for classification of the family Hepeviridae
- PMID: 24989172
- PMCID: PMC4165930
- DOI: 10.1099/vir.0.068429-0
Consensus proposals for classification of the family Hepeviridae
Erratum in
-
Consensus proposals for classification of the family Hepeviridae.J Gen Virol. 2015 May;96(Pt 5):1191-1192. doi: 10.1099/vir.0.000115. J Gen Virol. 2015. PMID: 26015322 Free PMC article. No abstract available.
Abstract
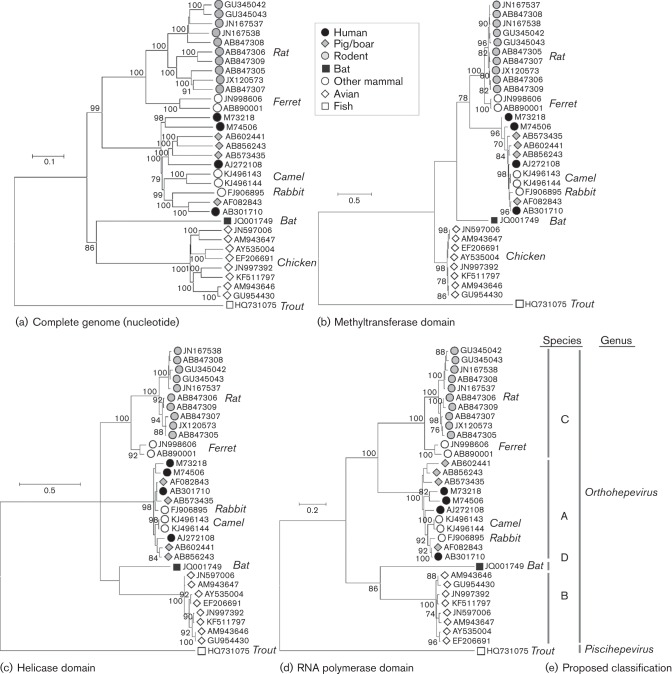
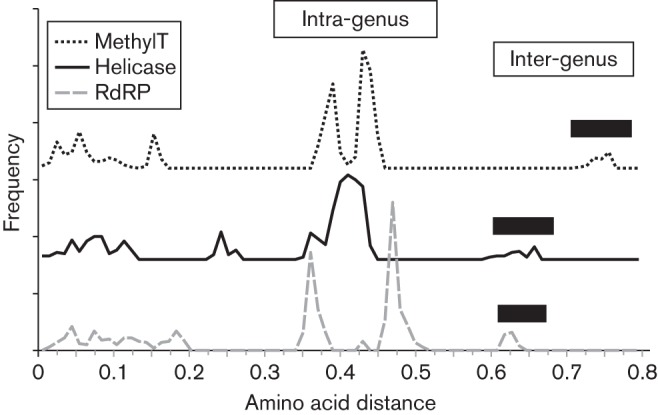
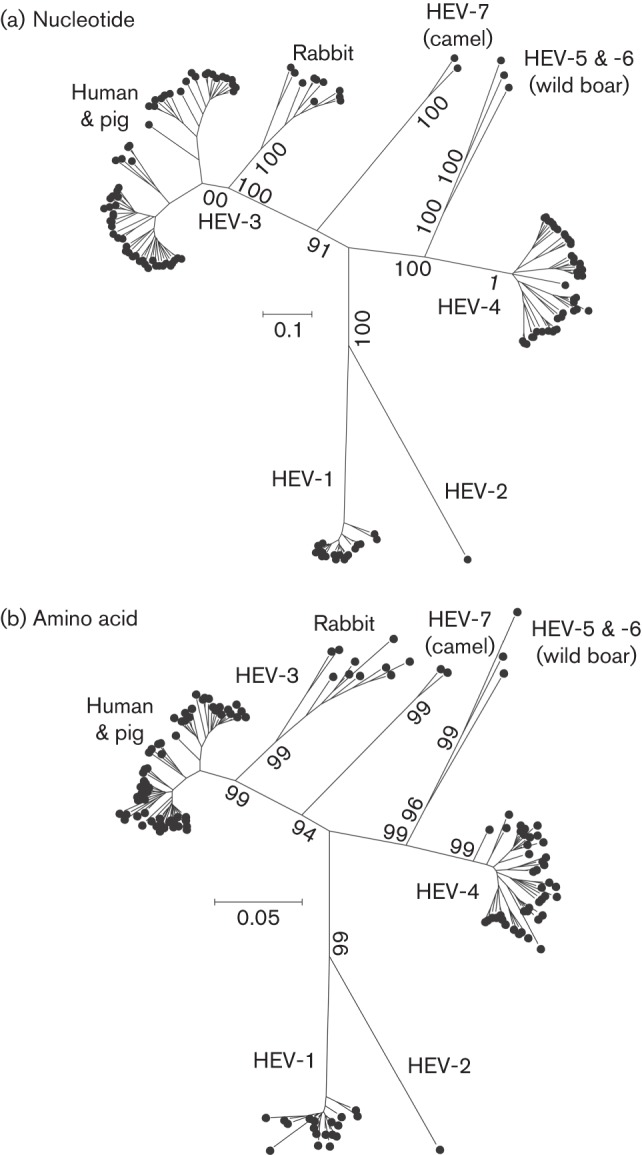
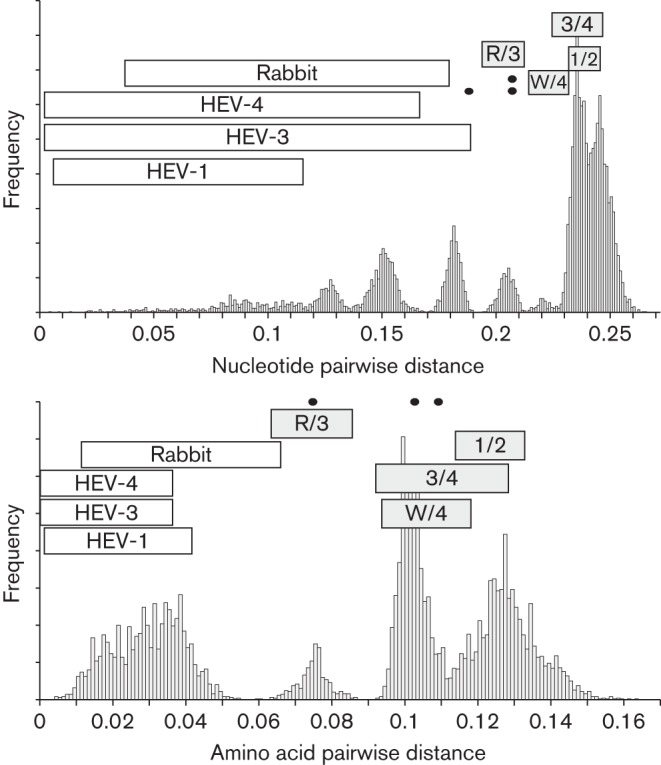
The family Hepeviridae consists of positive-stranded RNA viruses that infect a wide range of mammalian species, as well as chickens and trout. A subset of these viruses infects humans and can cause a self-limiting acute hepatitis that may become chronic in immunosuppressed individuals. Current published descriptions of the taxonomical divisions within the family Hepeviridae are contradictory in relation to the assignment of species and genotypes. Through analysis of existing sequence information, we propose a taxonomic scheme in which the family is divided into the genera Orthohepevirus (all mammalian and avian hepatitis E virus (HEV) isolates) and Piscihepevirus (cutthroat trout virus). Species within the genus Orthohepevirus are designated Orthohepevirus A (isolates from human, pig, wild boar, deer, mongoose, rabbit and camel), Orthohepevirus B (isolates from chicken), Orthohepevirus C (isolates from rat, greater bandicoot, Asian musk shrew, ferret and mink) and Orthohepevirus D (isolates from bat). Proposals are also made for the designation of genotypes within the human and rat HEVs. This hierarchical system is congruent with hepevirus phylogeny, and the three classification levels (genus, species and genotype) are consistent with, and reflect discontinuities in the ranges of pairwise distances between amino acid sequences. Adoption of this system would include the avoidance of host names in taxonomic identifiers and provide a logical framework for the assignment of novel variants.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases