

Osiris: accessible and reproducible phylogenetic and phylogenomic analyses within the Galaxy workflow management system
- PMID: 24990571
- PMCID: PMC4227113
- DOI: 10.1186/1471-2105-15-230
Osiris: accessible and reproducible phylogenetic and phylogenomic analyses within the Galaxy workflow management system
Abstract
Background: Phylogenetic tools and 'tree-thinking' approaches increasingly permeate all biological research. At the same time, phylogenetic data sets are expanding at breakneck pace, facilitated by increasingly economical sequencing technologies. Therefore, there is an urgent need for accessible, modular, and sharable tools for phylogenetic analysis.
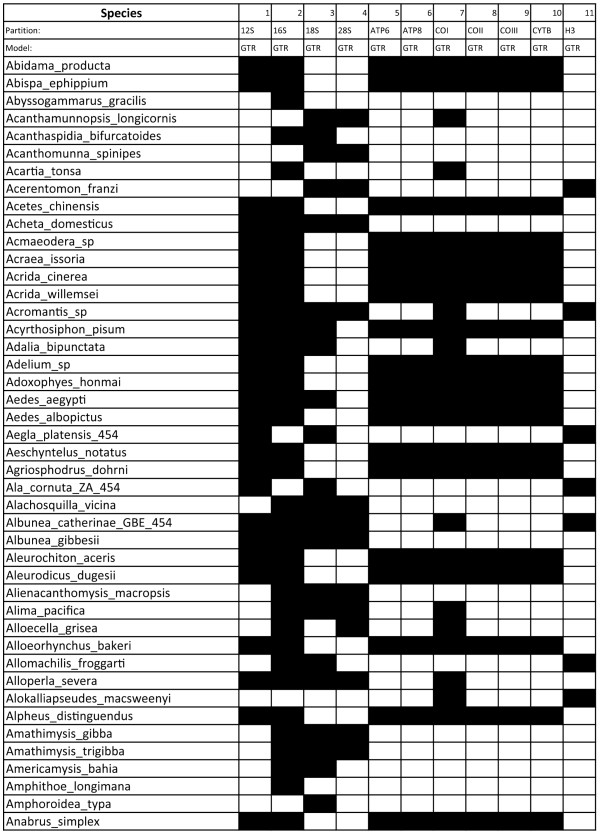
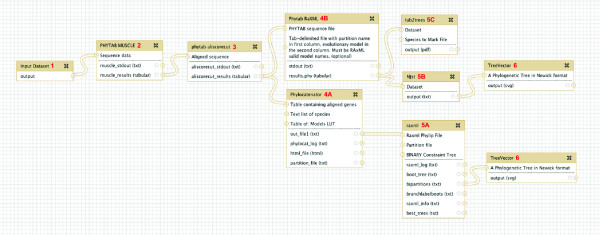
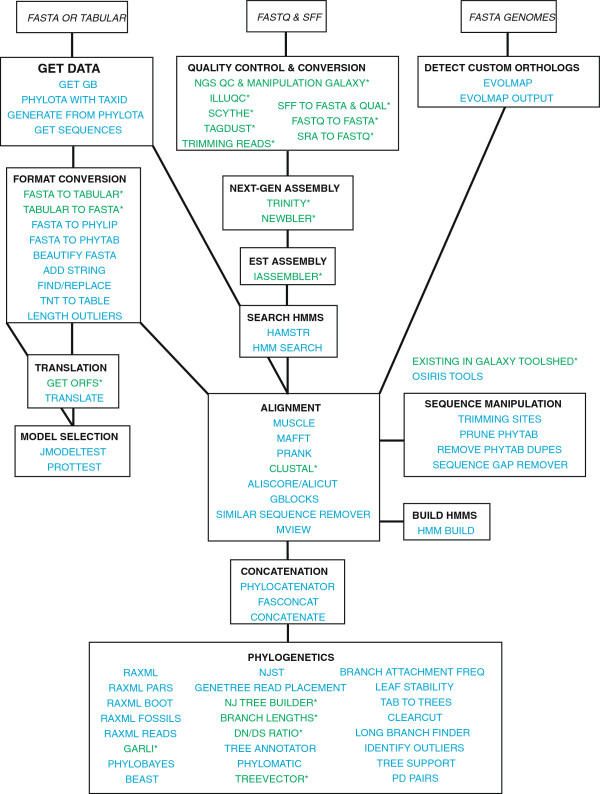
Results: We developed a suite of wrappers for new and existing phylogenetics tools for the Galaxy workflow management system that we call Osiris. Osiris and Galaxy provide a sharable, standardized, modular user interface, and the ability to easily create complex workflows using a graphical interface. Osiris enables all aspects of phylogenetic analysis within Galaxy, including de novo assembly of high throughput sequencing reads, ortholog identification, multiple sequence alignment, concatenation, phylogenetic tree estimation, and post-tree comparative analysis. The open source files are available on in the Bitbucket public repository and many of the tools are demonstrated on a public web server (http://galaxy-dev.cnsi.ucsb.edu/osiris/).
Conclusions: Osiris can serve as a foundation for other phylogenomic and phylogenetic tool development within the Galaxy platform.
Figures
References
-
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton A, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. pp. 1647–1649. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
