

Genetic inhibition of phosphorylation of the translation initiation factor eIF2α does not block Aβ-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer's disease
- PMID: 24992504
- PMCID: PMC4081565
- DOI: 10.1371/journal.pone.0101643
Genetic inhibition of phosphorylation of the translation initiation factor eIF2α does not block Aβ-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer's disease
Erratum in
- PLoS One. 2014;9(10):e110914
Abstract
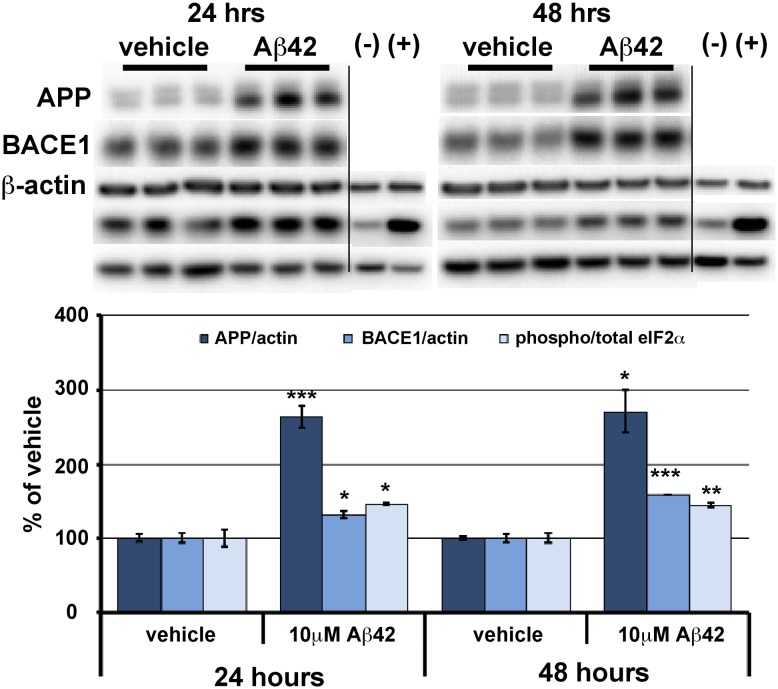
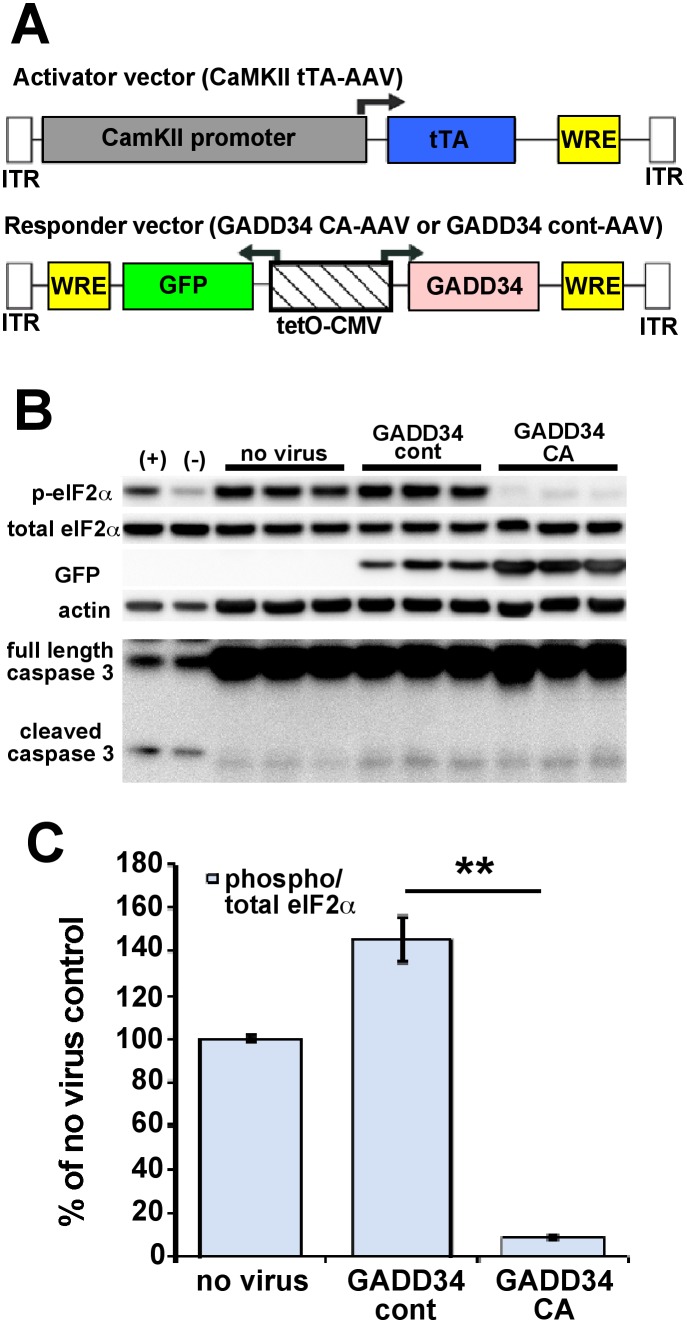
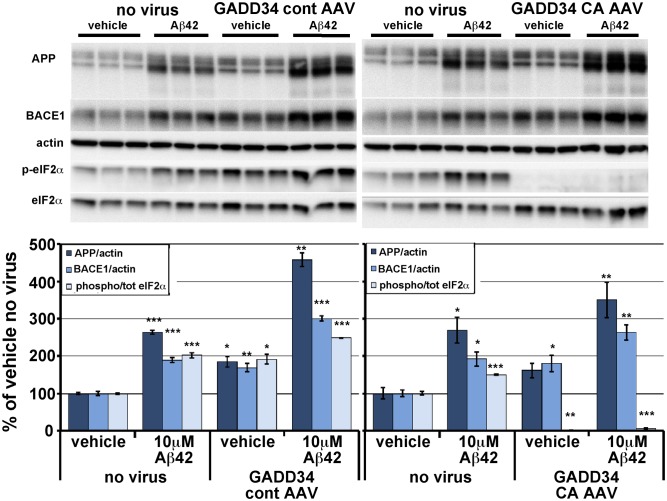
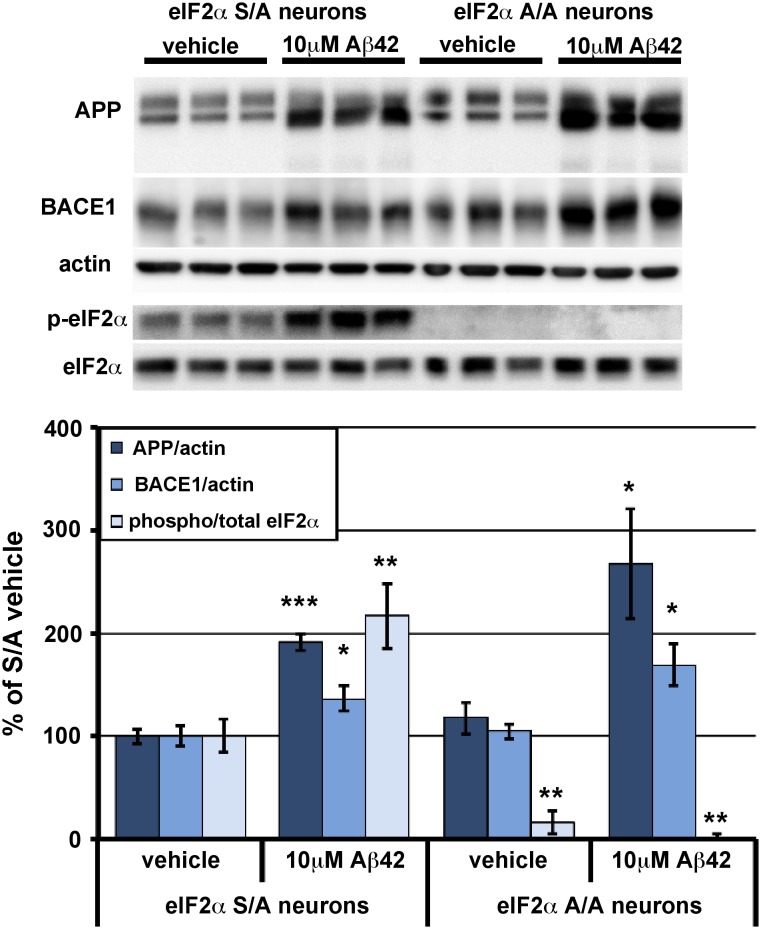
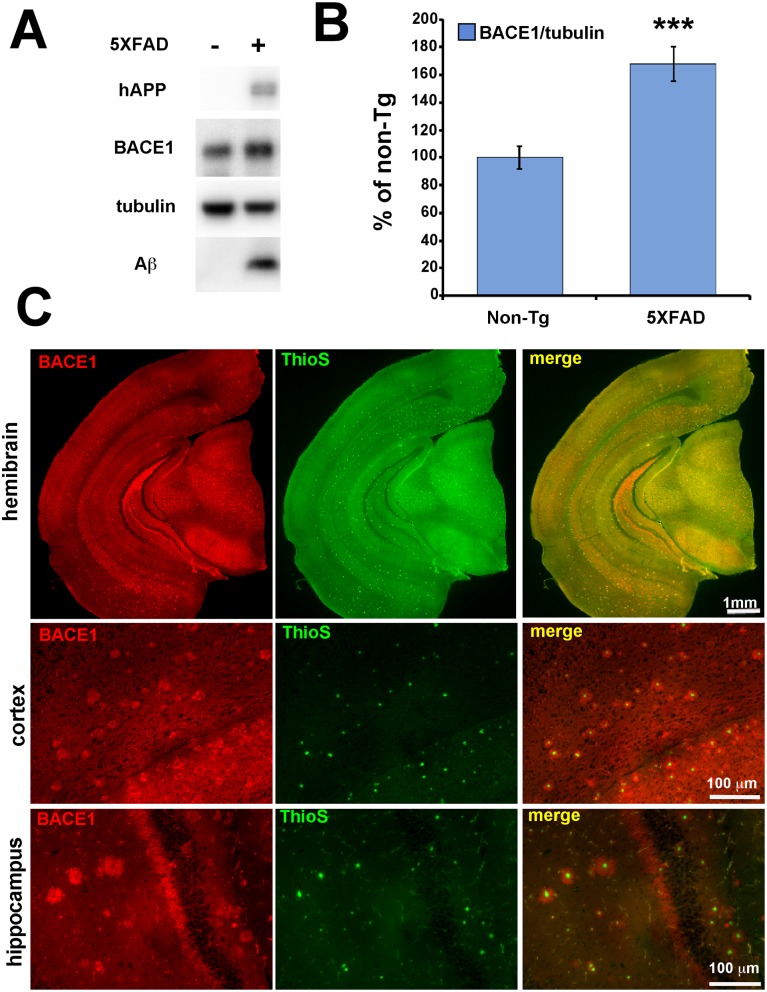
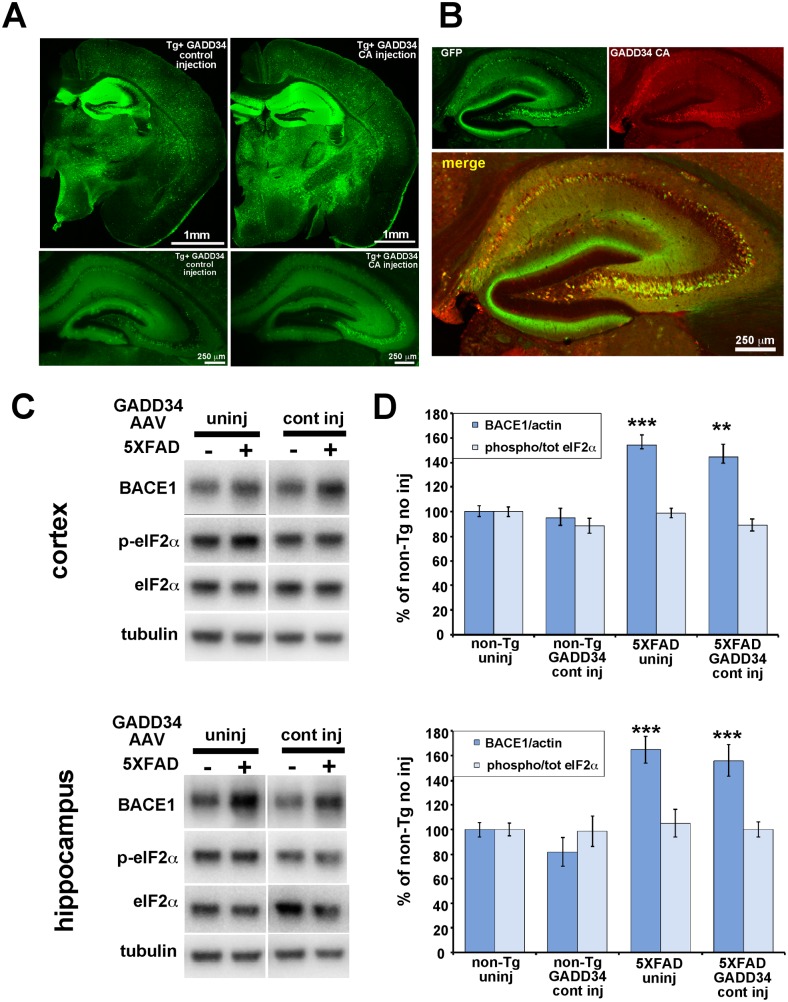
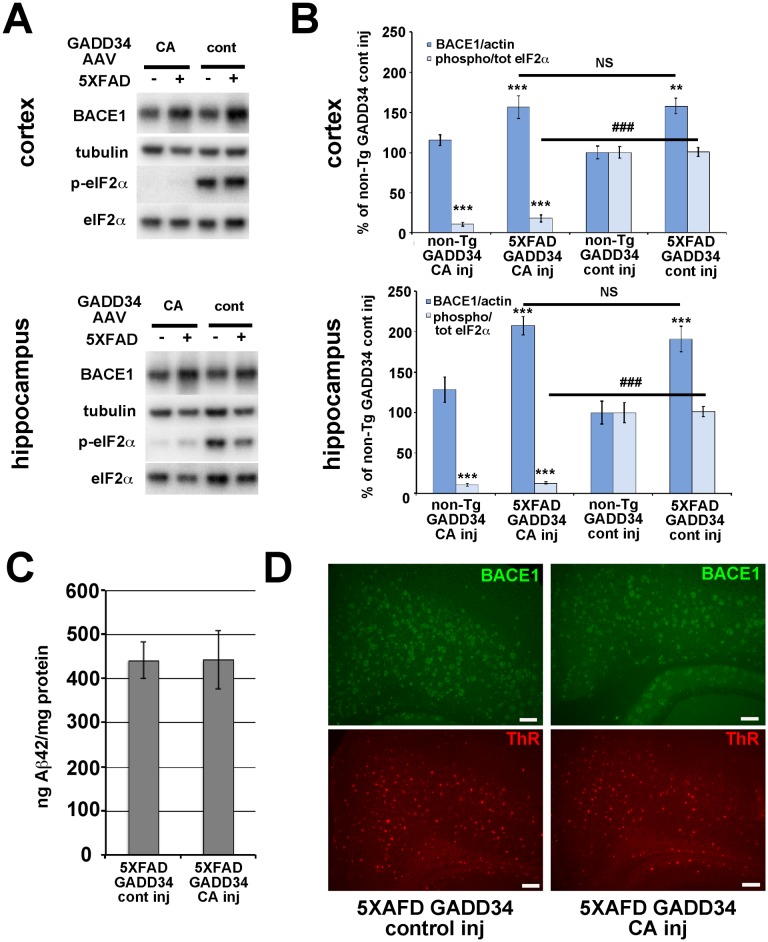
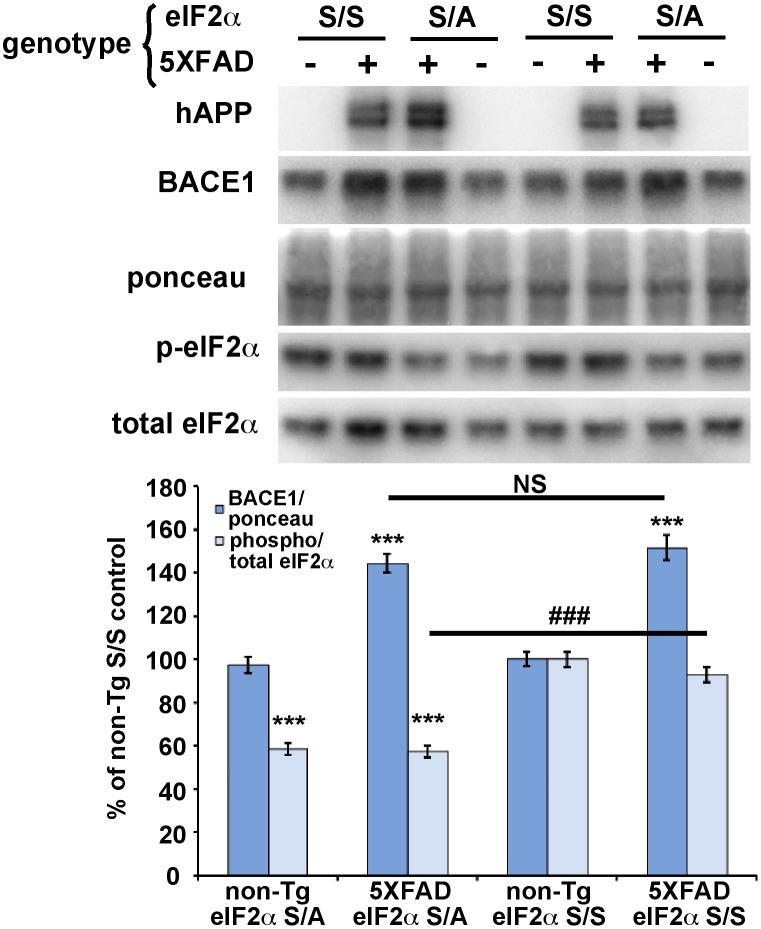
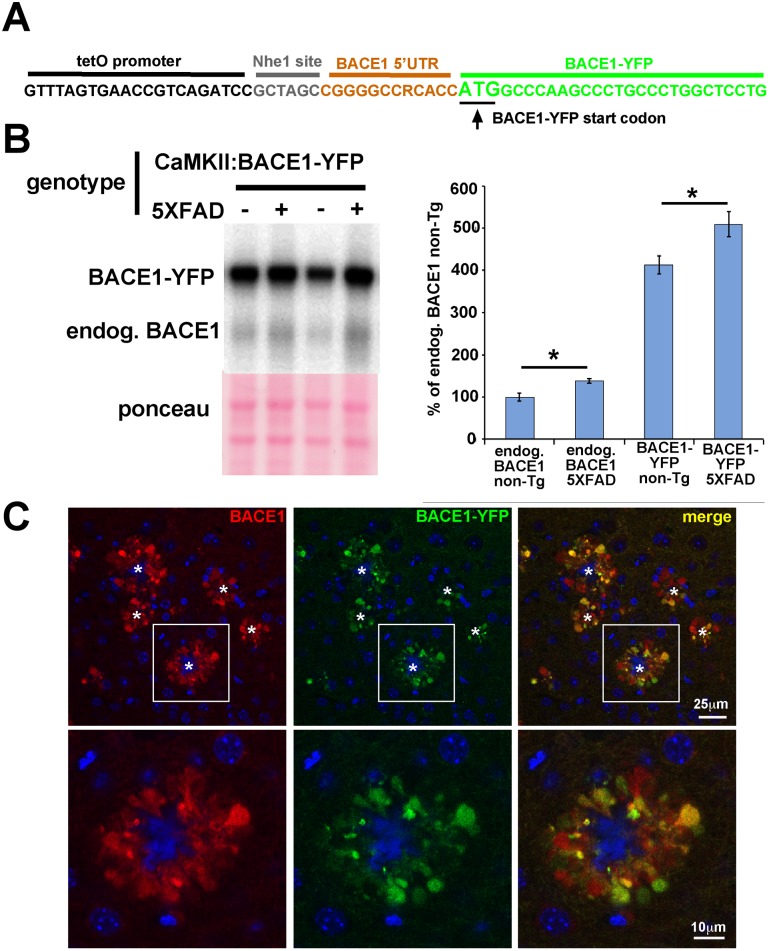
β-site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) initiates the production of β-amyloid (Aβ), the major constituent of amyloid plaques in Alzheimer's disease (AD). BACE1 is elevated ∼2-3 fold in AD brain and is concentrated in dystrophic neurites near plaques, suggesting BACE1 elevation is Aβ-dependent. Previously, we showed that phosphorylation of the translation initiation factor eIF2α de-represses translation of BACE1 mRNA following stress such as energy deprivation. We hypothesized that stress induced by Aβ might increase BACE1 levels by the same translational mechanism involving eIF2α phosphorylation. To test this hypothesis, we used three different genetic strategies to determine the effects of reducing eIF2α phosphorylation on Aβ-dependent BACE1 elevation in vitro and in vivo: 1) a two-vector adeno-associated virus (AAV) system to express constitutively active GADD34, the regulatory subunit of PP1c eIF2α phosphatase; 2) a non-phosphorylatable eIF2α S51A knockin mutation; 3) a BACE1-YFP transgene lacking the BACE1 mRNA 5' untranslated region (UTR) required for eIF2α translational regulation. The first two strategies were used in primary neurons and 5XFAD transgenic mice, while the third strategy was employed only in 5XFAD mice. Despite very effective reduction of eIF2α phosphorylation in both primary neurons and 5XFAD brains, or elimination of eIF2α-mediated regulation of BACE1-YFP mRNA translation in 5XFAD brains, Aβ-dependent BACE1 elevation was not decreased. Additionally, robust inhibition of eIF2α phosphorylation did not block Aβ-dependent APP elevation in primary neurons, nor did it reduce amyloid pathology in 5XFAD mice. We conclude that amyloid-associated BACE1 elevation is not caused by translational de-repression via eIF2α phosphorylation, but instead appears to involve a post-translational mechanism. These definitive genetic results exclude a role for eIF2α phosphorylation in Aβ-dependent BACE1 and APP elevation. We suggest a vicious pathogenic cycle wherein Aβ42 toxicity induces peri-plaque BACE1 and APP accumulation in dystrophic neurites leading to exacerbated Aβ production and plaque progression.
Conflict of interest statement
Figures
References
-
- Sisodia SS, St George-Hyslop PH (2002) gamma-Secretase, Notch, Abeta and Alzheimer’s disease: where do the presenilins fit in? Nat Rev Neurosci 3: 281–290. - PubMed
-
- Klein WL, Stine WB Jr, Teplow DB (2004) Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging 25: 569–580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DK088227/DK/NIDDK NIH HHS/United States
- DK042394/DK/NIDDK NIH HHS/United States
- F32AG033445/AG/NIA NIH HHS/United States
- HL052173/HL/NHLBI NIH HHS/United States
- R37 DK042394/DK/NIDDK NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- R01 AG030142/AG/NIA NIH HHS/United States
- 5T32AG00026/AG/NIA NIH HHS/United States
- CA060553/CA/NCI NIH HHS/United States
- F32 AG033445/AG/NIA NIH HHS/United States
- R01 DK088227/DK/NIDDK NIH HHS/United States
- R01 DK042394/DK/NIDDK NIH HHS/United States
- R01 HL052173/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
