

The archaeal legacy of eukaryotes: a phylogenomic perspective
- PMID: 24993577
- PMCID: PMC4176008
- DOI: 10.1101/cshperspect.a016022
The archaeal legacy of eukaryotes: a phylogenomic perspective
Abstract
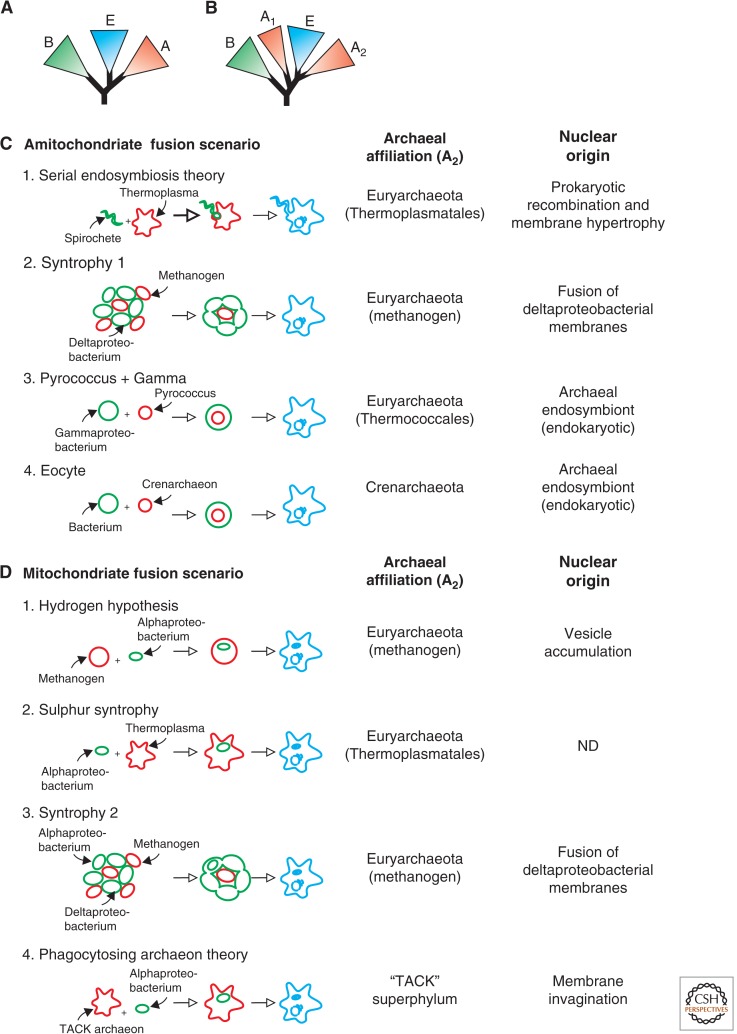
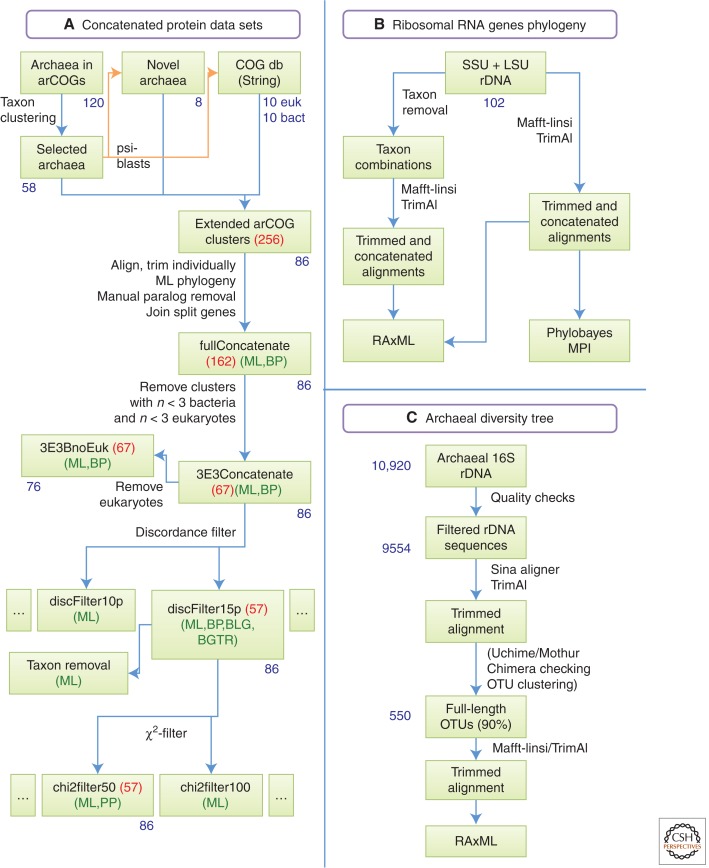
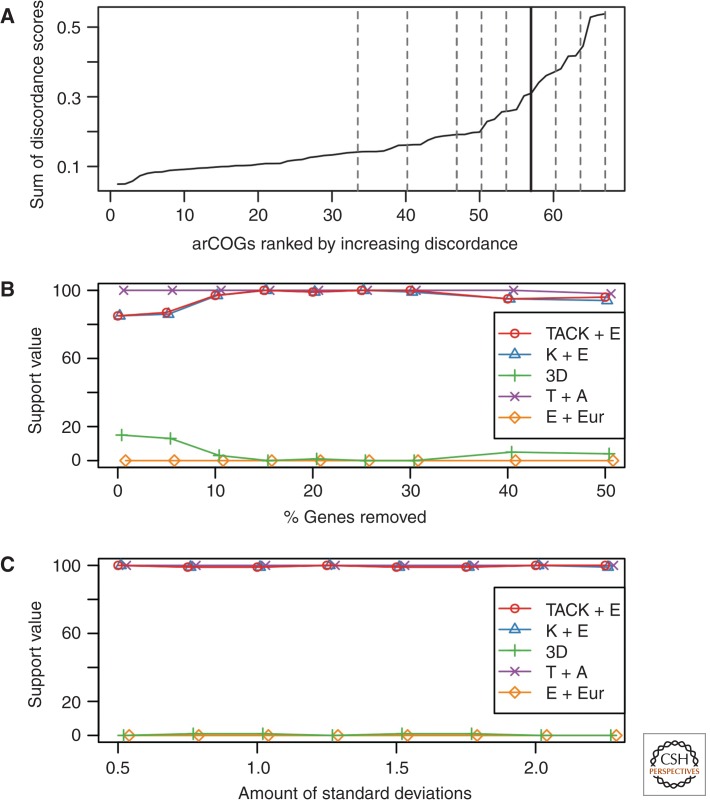
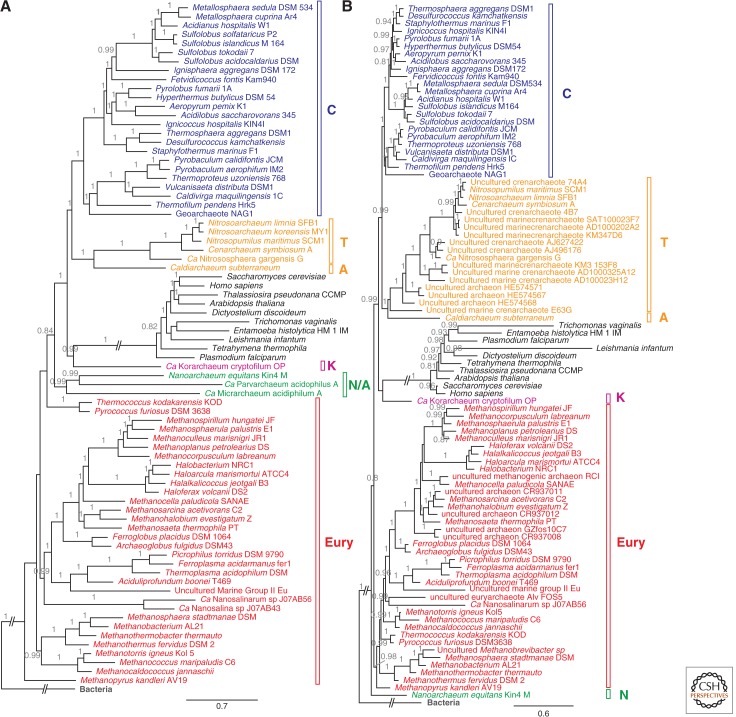
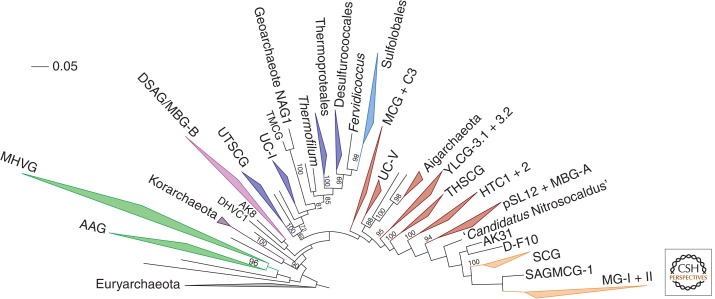
The origin of the eukaryotic cell can be regarded as one of the hallmarks in the history of life on our planet. The apparent genomic chimerism in eukaryotic genomes is currently best explained by invoking a cellular fusion at the root of the eukaryotes that involves one archaeal and one or more bacterial components. Here, we use a phylogenomics approach to reevaluate the evolutionary affiliation between Archaea and eukaryotes, and provide further support for scenarios in which the nuclear lineage in eukaryotes emerged from within the archaeal radiation, displaying a strong phylogenetic affiliation with, or even within, the archaeal TACK superphylum. Further taxonomic sampling of archaeal genomes in this superphylum will certainly provide a better resolution in the events that have been instrumental for the emergence of the eukaryotic lineage.
Copyright © 2014 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Bell PJ 2001. Viral eukaryogenesis: Was the ancestor of the nucleus a complex DNA virus? J Mol Evol 53: 251–256 - PubMed
-
- Bult CJ, White O, Olsen GJ, Zhou L, Fleischmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD, et al. 1996. Complete genome sequence of the methanogenic archaeon, Methanococcus jannaschii. Science 273: 1058–1073 - PubMed
-
- Cavalier-Smith T 1989. Molecular phylogeny. Archaebacteria and Archezoa. Nature 339: l00–101 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources