

Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors
- PMID: 24995006
- PMCID: PMC4062968
- DOI: 10.3389/fimmu.2014.00269
Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors
Abstract
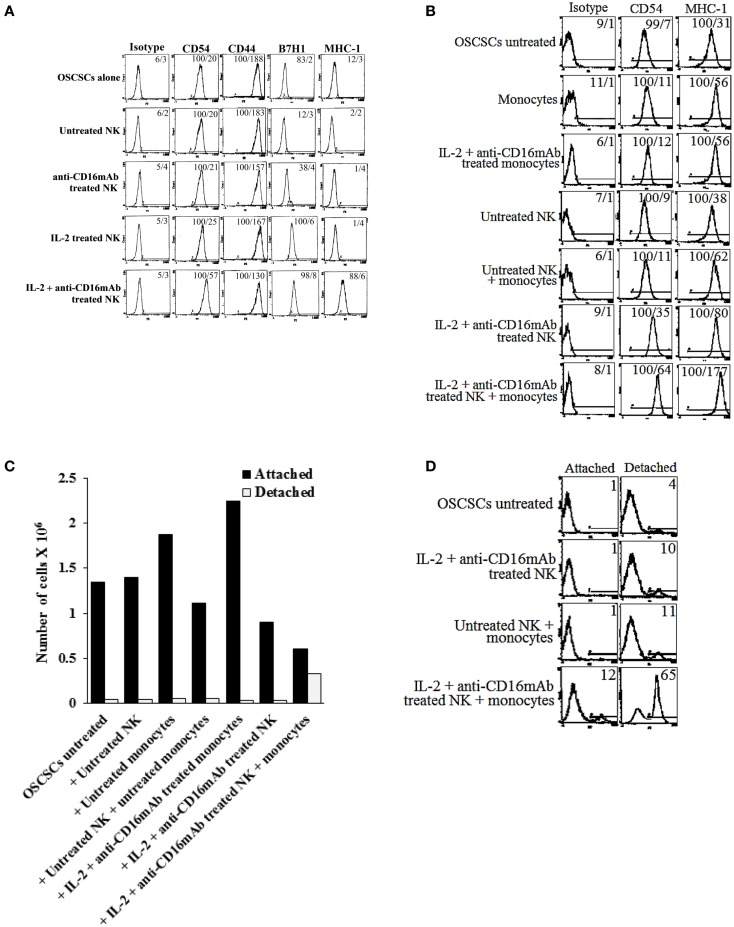
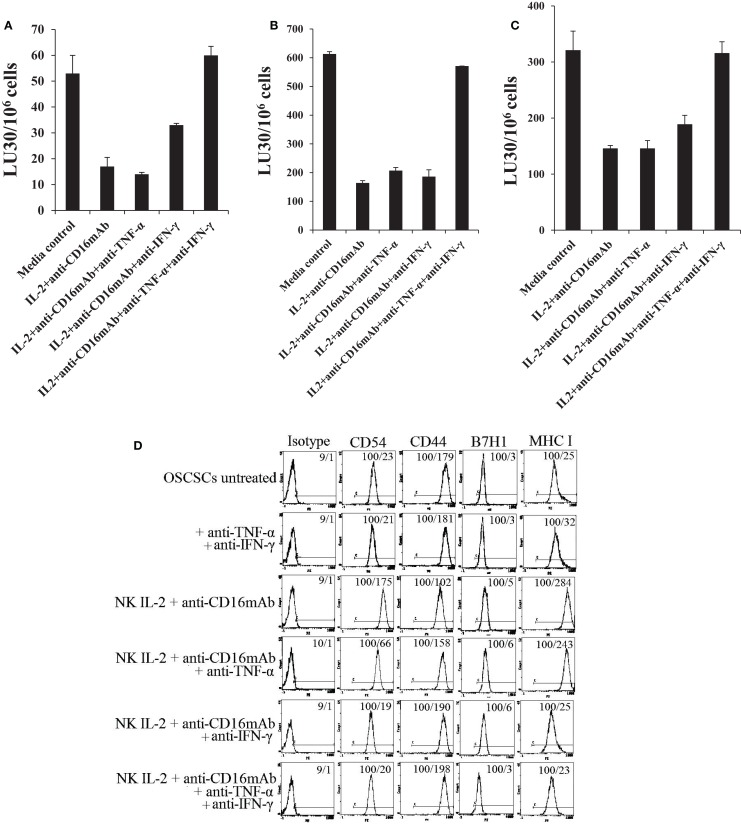
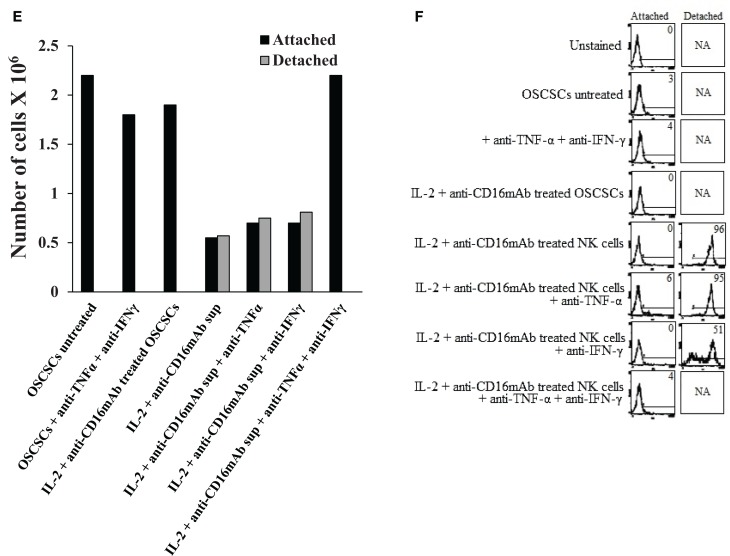
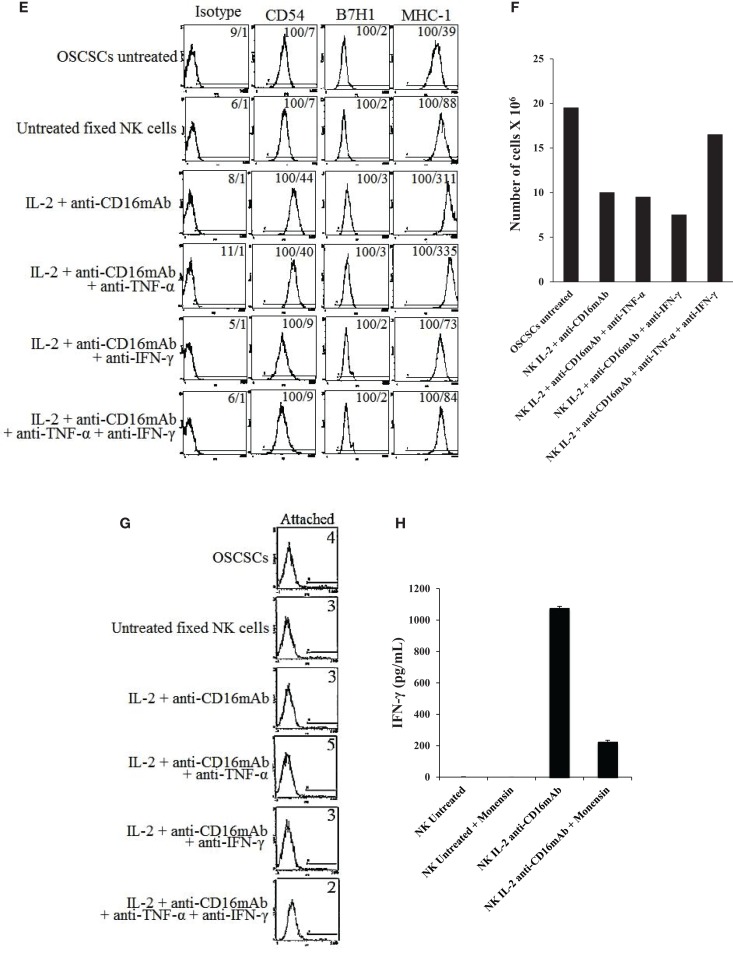
In this paper, we provide evidence that anergized NK cells through secreted factors and direct cell-cell contact have the ability to induce differentiation of healthy dental pulp stem cells and stem cell of apical papillae as well as transformed oral squamous cancer stem cell (OSCSC) and Mia-Paca-2, poorly differentiated stem-like pancreatic tumors, resulting in their resistance to NK cell-mediated cytotoxicity. Induction of NK cell resistance and differentiation in the stem cells correlated with the increased expression of CD54, B7H1, and MHC class I, and mediated by the combination of membrane-bound or secreted IFN-γ and TNF-α from the NK cells since antibodies to both cytokines and not each one alone were able to inhibit differentiation or resistance to NK cells. Similarly, antibodies to both TNF-α and IFN-γ were required to prevent NK-mediated inhibition of cell growth, and restored the numbers of the stem cells to the levels obtained when stem cells were cultured in the absence of anergized NK cells. Interestingly, the effect of anti-IFN-γ antibody in the absence of anti-TNF-α antibody was more dominant for the prevention of increase in surface receptor expression since its addition abrogated the increase in CD54, B7H1, and MHC class I surface expression. Antibodies to CD54 or LFA-1 was unable to inhibit differentiation whereas antibodies to MHC class I but not B7H1 increased cytotoxicity of well-differentiated oral squamous carcinoma cells as well as OSCSCs differentiated by the IL-2 + anti-CD16 mAb-treated NK cells whereas it inhibited the cytotoxicity of NK cells against OSCSCs. Thus, NK cells may inhibit the progression of cancer by killing and/or differentiation of cancer stem cells, which severely halt cancer growth, invasion, and metastasis.
Keywords: IFN-γ; MP2; NK; OSCCs; OSCSCs; cytotoxicity; regulatory NK.
Figures












References
-
- Kolenko V, Wang Q, Riedy MC, O’Shea J, Ritz J, Cathcart MK, et al. Tumor-induced suppression of T lymphocyte proliferation coincides with inhibition of Jak3 expression and IL-2 receptor signaling: role of soluble products from human renal cell carcinomas. J Immunol (1997) 159(6):3057–67 - PubMed
-
- Bennett MW, O’Connell J, O’Sullivan GC, Brady C, Roche D, Collins JK, et al. The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J Immunol (1998) 160(11):5669–75 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials