

Expansion of biological pathways based on evolutionary inference
- PMID: 24995987
- PMCID: PMC4171950
- DOI: 10.1016/j.cell.2014.05.034
Expansion of biological pathways based on evolutionary inference
Abstract
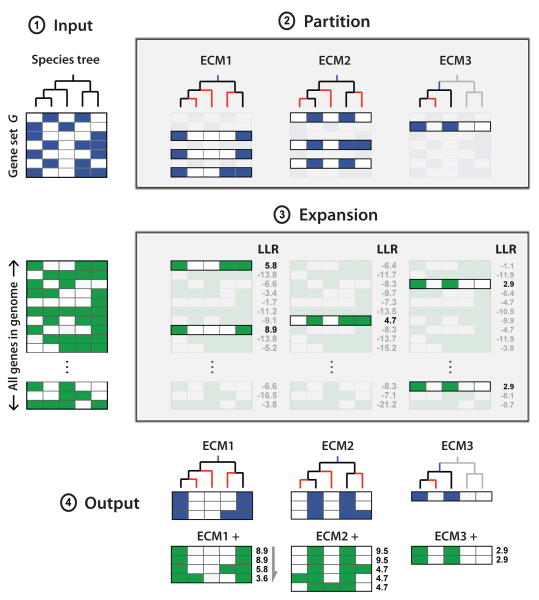
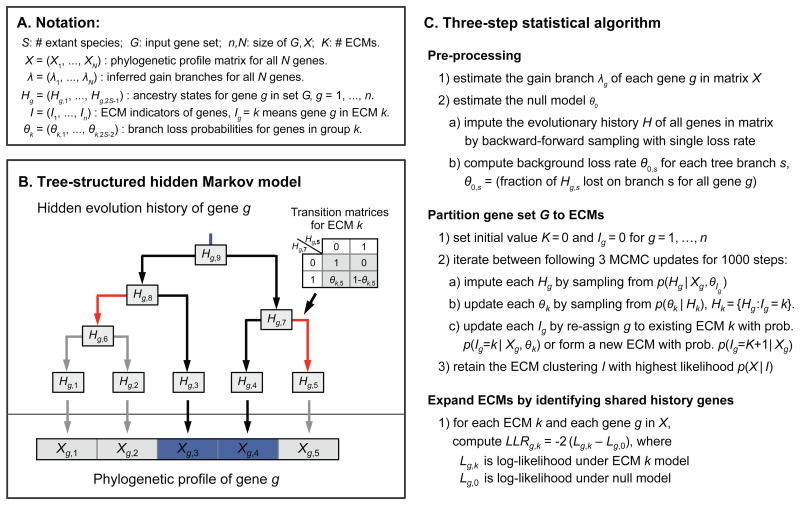
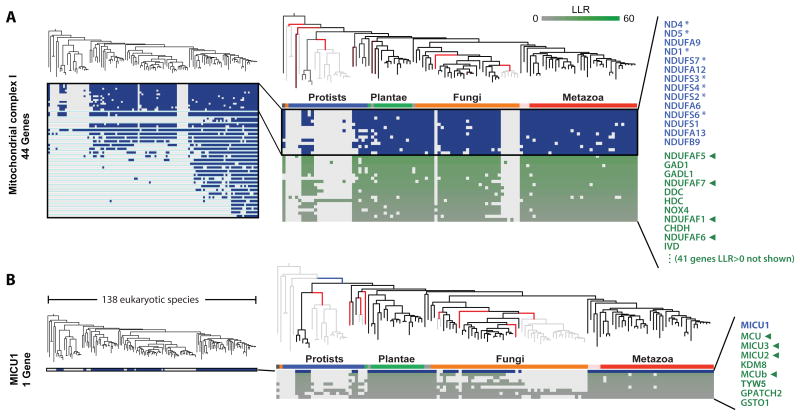
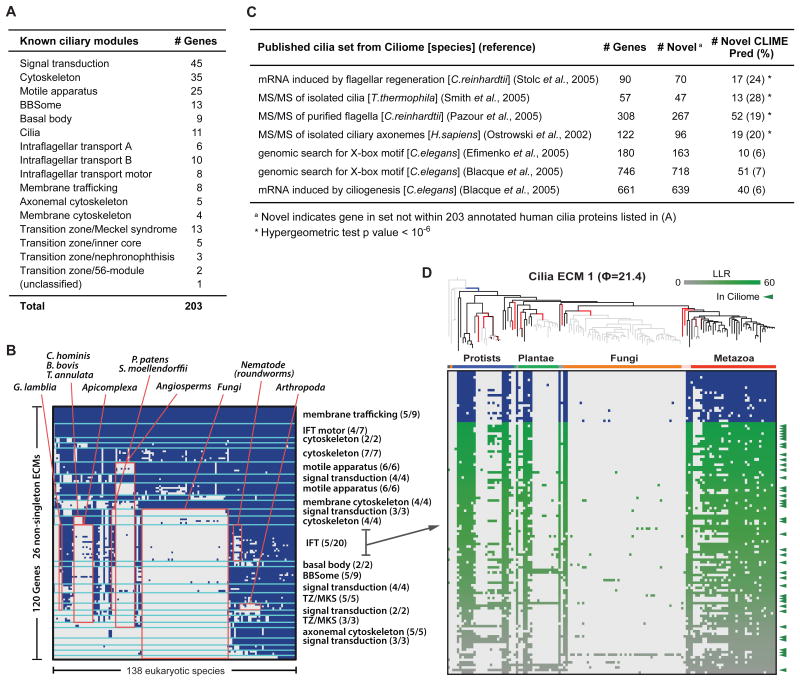
The availability of diverse genomes makes it possible to predict gene function based on shared evolutionary history. This approach can be challenging, however, for pathways whose components do not exhibit a shared history but rather consist of distinct "evolutionary modules." We introduce a computational algorithm, clustering by inferred models of evolution (CLIME), which inputs a eukaryotic species tree, homology matrix, and pathway (gene set) of interest. CLIME partitions the gene set into disjoint evolutionary modules, simultaneously learning the number of modules and a tree-based evolutionary history that defines each module. CLIME then expands each module by scanning the genome for new components that likely arose under the inferred evolutionary model. Application of CLIME to ∼1,000 annotated human pathways and to the proteomes of yeast, red algae, and malaria reveals unanticipated evolutionary modularity and coevolving components. CLIME is freely available and should become increasingly powerful with the growing wealth of eukaryotic genomes.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
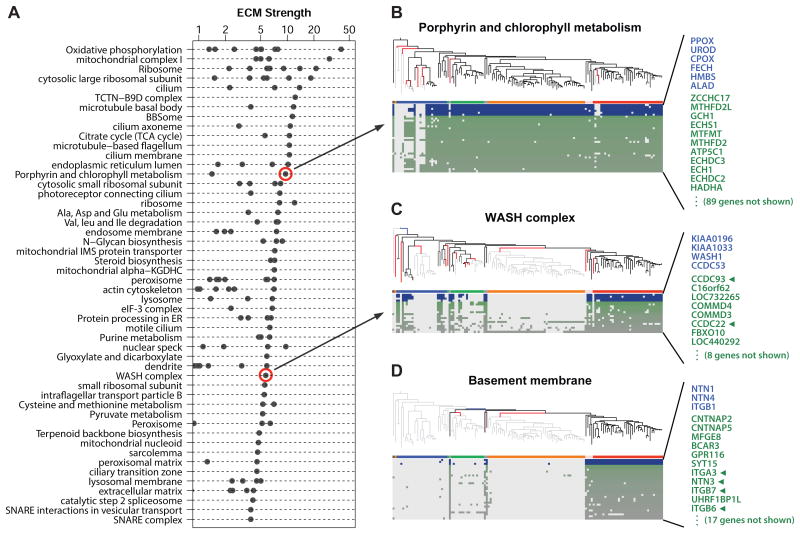
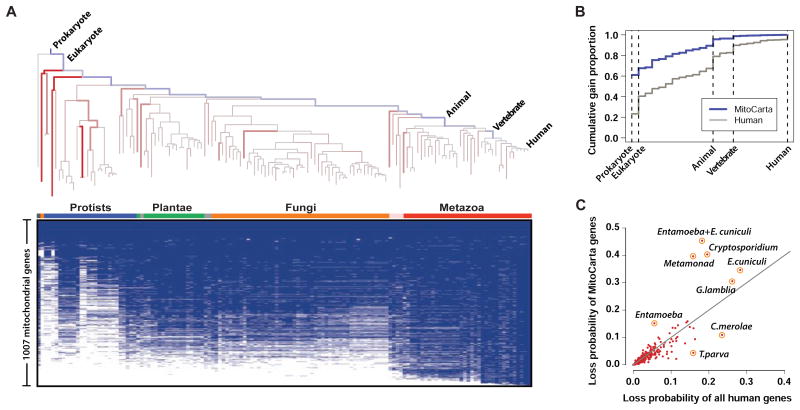
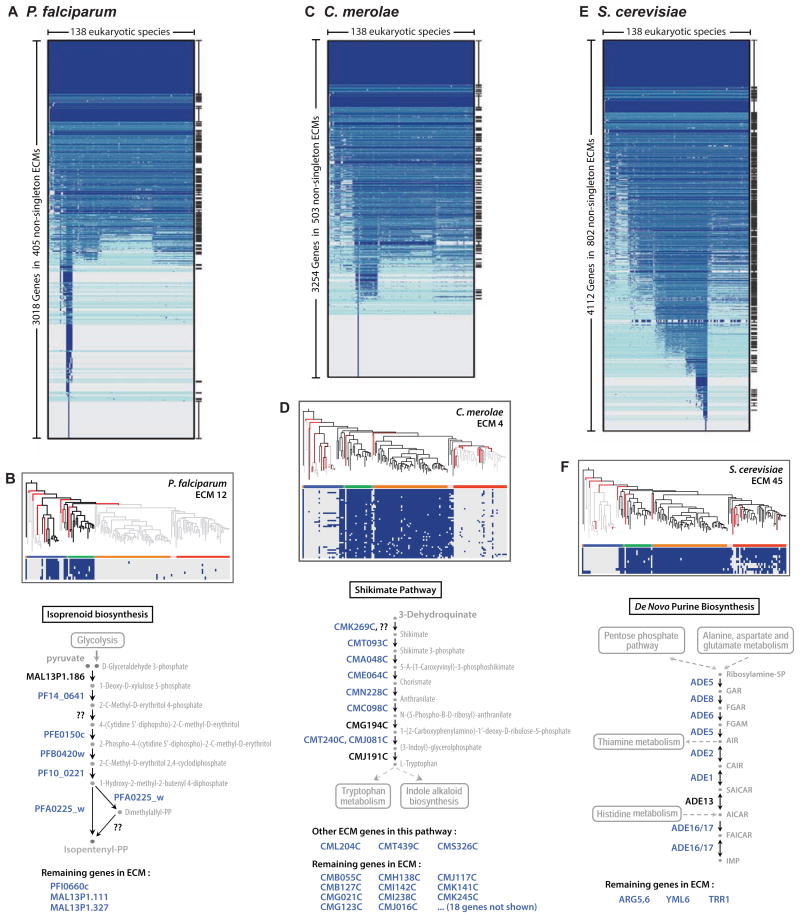
Comment in
-
Finding a common path: predicting gene function using inferred evolutionary trees.Dev Cell. 2014 Jul 14;30(1):4-5. doi: 10.1016/j.devcel.2014.06.029. Dev Cell. 2014. PMID: 25026031
References
-
- Balsa E, Marco R, Perales-Clemente E, Szklarczyk R, Calvo E, Landázuri MO, Enríquez JA. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metabolism. 2012;16:378–386. - PubMed
-
- Barker D, Meade A, Pagel M. Constrained models of evolution lead to improved prediction of functional linkage from correlated gain and loss of genes. Bioinformatics. 2007;23:14–20. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
