

The 2013 SFRBM discovery award: selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment
- PMID: 24996204
- PMCID: PMC4146642
- DOI: 10.1016/j.freeradbiomed.2014.06.006
The 2013 SFRBM discovery award: selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment
Abstract
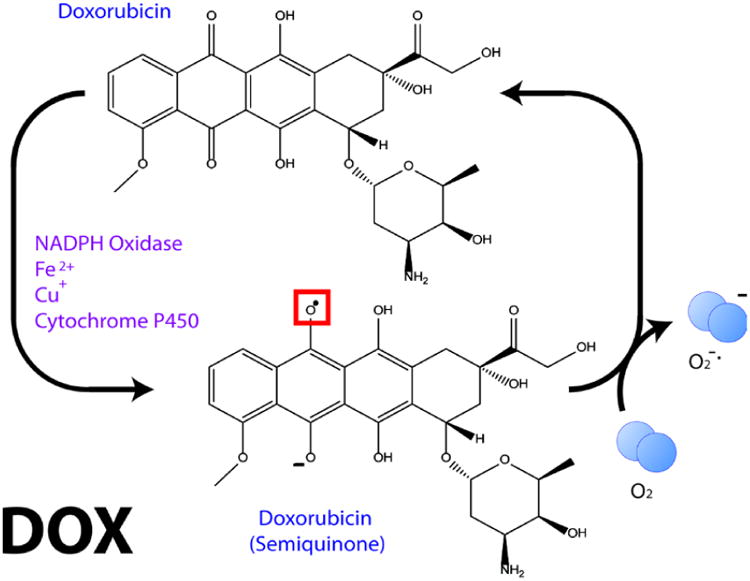
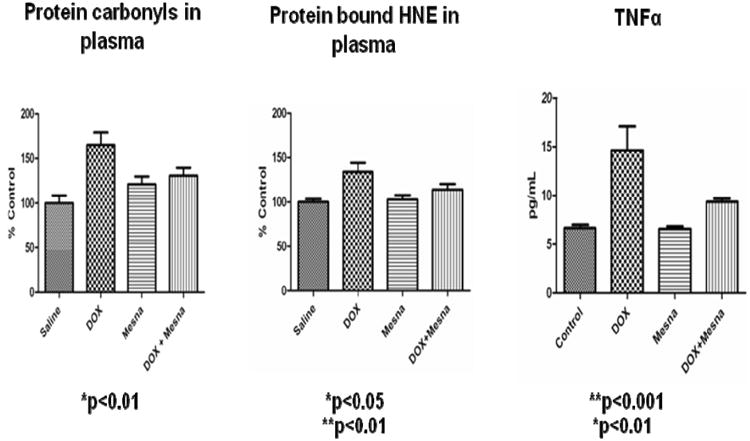
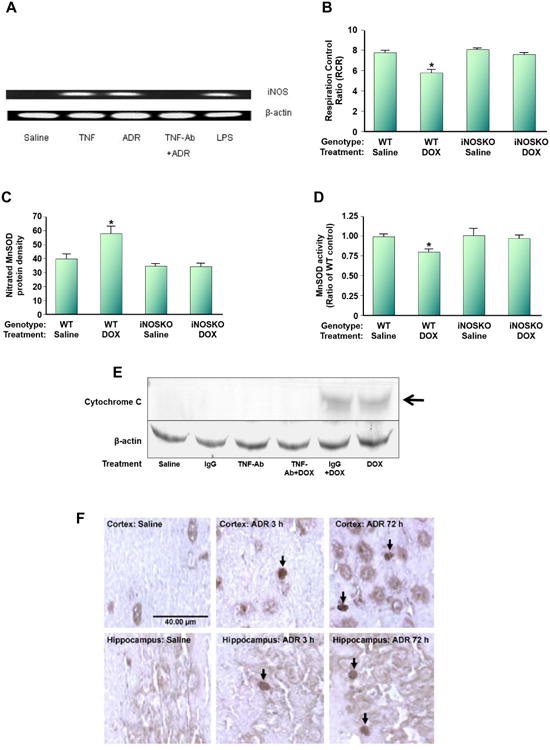
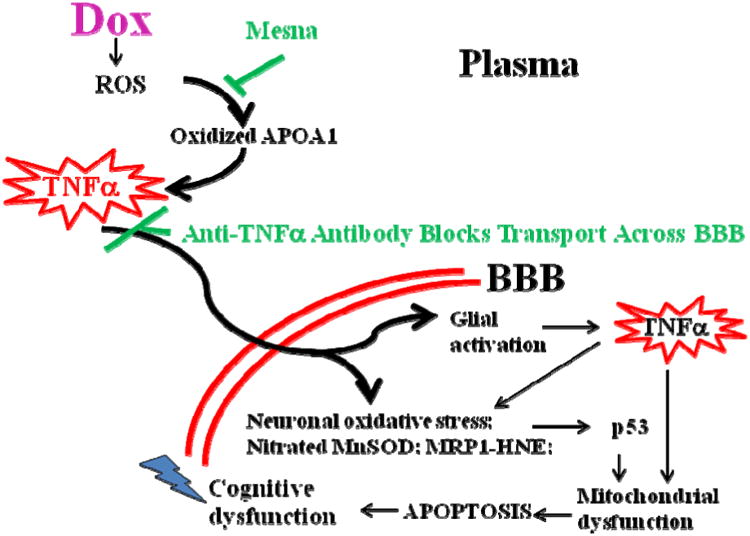
This retrospective review on discoveries of the roles of oxidative stress in brain of subjects with Alzheimer disease (AD) and animal models thereof as well as brain from animal models of chemotherapy-induced cognitive impairment (CICI) results from the author receiving the 2013 Discovery Award from the Society for Free Radical Biology and Medicine. The paper reviews our laboratory's discovery of protein oxidation and lipid peroxidation in AD brain regions rich in amyloid β-peptide (Aβ) but not in Aβ-poor cerebellum; redox proteomics as a means to identify oxidatively modified brain proteins in AD and its earlier forms that are consistent with the pathology, biochemistry, and clinical presentation of these disorders; how Aβ in in vivo, ex vivo, and in vitro studies can lead to oxidative modification of key proteins that also are oxidatively modified in AD brain; the role of the single methionine residue of Aβ(1-42) in these processes; and some of the potential mechanisms in the pathogenesis and progression of AD. CICI affects a significant fraction of the 14 million American cancer survivors, and due to diminished cognitive function, reduced quality of life of the persons with CICI (called "chemobrain" by patients) often results. A proposed mechanism for CICI employed the prototypical ROS-generating and non-blood brain barrier (BBB)-penetrating chemotherapeutic agent doxorubicin (Dox, also called adriamycin, ADR). Because of the quinone moiety within the structure of Dox, this agent undergoes redox cycling to produce superoxide free radical peripherally. This, in turn, leads to oxidative modification of the key plasma protein, apolipoprotein A1 (ApoA1). Oxidized ApoA1 leads to elevated peripheral TNFα, a proinflammatory cytokine that crosses the BBB to induce oxidative stress in brain parenchyma that affects negatively brain mitochondria. This subsequently leads to apoptotic cell death resulting in CICI. This review outlines aspects of CICI consistent with the clinical presentation, biochemistry, and pathology of this disorder. To the author's knowledge this is the only plausible and self-consistent mechanism to explain CICI. These two different disorders of the CNS affect millions of persons worldwide. Both AD and CICI share free radical-mediated oxidative stress in brain, but the source of oxidative stress is not the same. Continued research is necessary to better understand both AD and CICI. The discoveries about these disorders from the Butterfield Laboratory that led to the 2013 Discovery Award from the Society of Free Radical and Medicine provide a significant foundation from which this future research can be launched.
Keywords: 2013 SFRBM Discovery Award; Alzheimer disease (AD) and its earlier forms (amnestic MCI and preclinical AD); Aβ(1–42) associated oxidative stress; Chemotherapy-induced cognitive impairment (“chemobrain”); Plasma-derived elevated TNFα and its sequela in brain; Redox proteomics.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures

References
-
- Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. Oxford University Press; 2007. p. 704.
-
- Butterfield DA, Stadman ER. Protein oxidation processes in aging brain. Advances in Cell Aging and Gerontology. 1997;2:161–191.
-
- Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods in enzymology. 1994;233:229–240. - PubMed
-
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
