

γH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments
- PMID: 24996846
- PMCID: PMC4094550
- DOI: 10.1186/1471-2407-14-483
γH2AX and Chk1 phosphorylation as predictive pharmacodynamic biomarkers of Chk1 inhibitor-chemotherapy combination treatments
Abstract
Background: Chk1 inhibitors are currently in clinical trials in combination with a range of cytotoxic agents and have the potential to potentiate the clinical activity of a large number of standard of care chemotherapeutic agents. Utilizing pharmacodynamic biomarkers to optimize drug dose and scheduling in these trials could greatly enhance the likelihood of clinical success.
Methods: In this study, we evaluated the in vitro potentiation of the cytotoxicity of a range of cytotoxic chemotherapeutic drugs by the novel Chk1 inhibitor V158411 in p53 mutant colon cancer cells. Pharmacodynamic biomarkers were evaluated in vitro.
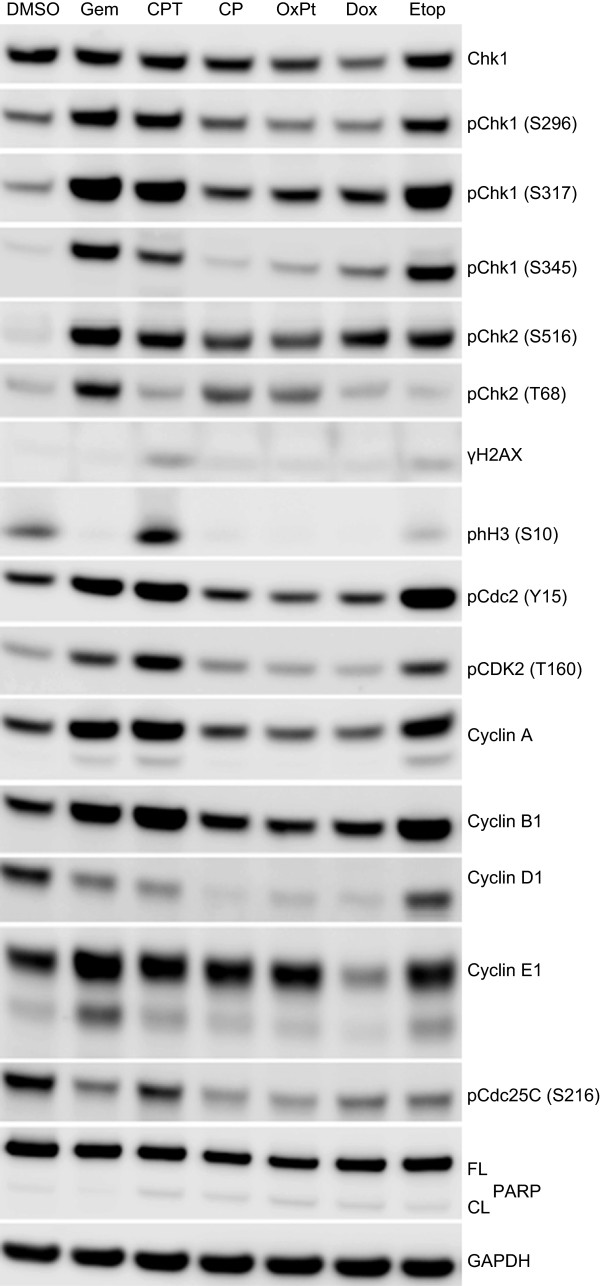
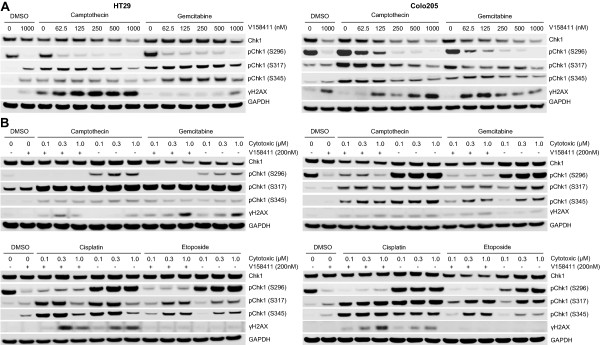
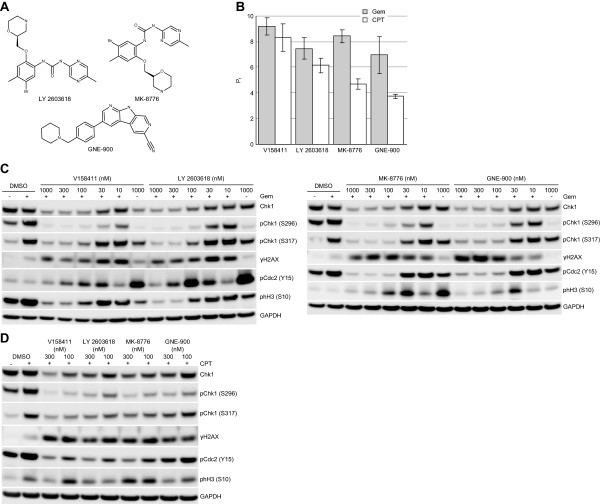
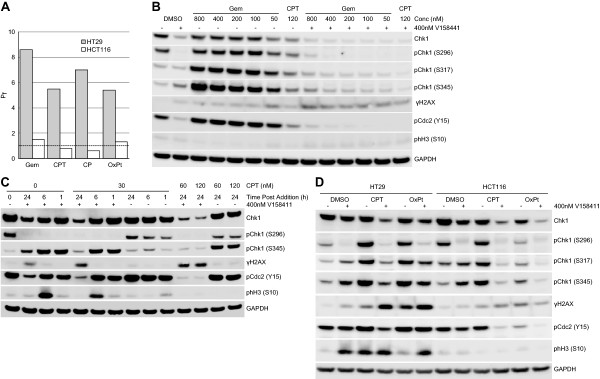
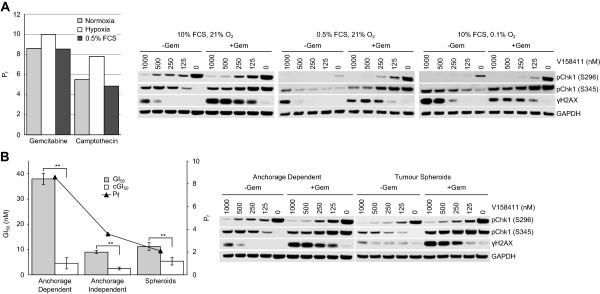
Results: V158411 potentiated the cytotoxicity of a range of chemotherapeutic agents with distinct mechanisms of action in p53 mutant colon cancer cell lines grown in anchorage dependent or independent culture conditions. Analysis of pharmacodynamic biomarker changes identified dependencies on the chemotherapeutic agent, the concentration of the chemotherapeutic and the duration of time between combination treatment and biomarker analysis. A reduction in total Chk1 and S296/S317/S345 phosphorylation occurred consistently with all cytotoxics in combination with V158411 but did not predict cell line potentiation. Induction of γH2AX levels was chemotherapeutic dependent and correlated closely with potentiation of gemcitabine and camptothecin in p53 mutant colon cancer cells.
Conclusions: Our results suggest that Chk1 phosphorylation could be a useful biomarker for monitoring inhibition of Chk1 activity in clinical trials involving a range of V158411-chemotherapy combinations and γH2AX induction as a predictor of potentiation in combinations containing gemcitabine or camptothecin.
Figures



References
-
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
