

Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11
- PMID: 24999486
- PMCID: PMC4078876
- DOI: 10.1002/acn3.64
Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11
Abstract
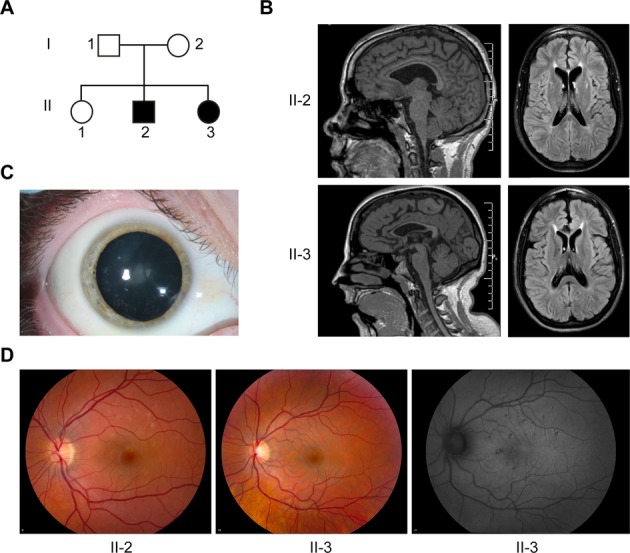
Objective: Hereditary spastic paraplegias (HSPs) are among the most genetically diverse inherited neurological disorders, with over 70 disease loci identified (SPG1-71) to date. SPG15 and SPG11 are clinically similar, autosomal recessive disorders characterized by progressive spastic paraplegia along with thin corpus callosum, white matter abnormalities, cognitive impairment, and ophthalmologic abnormalities. Furthermore, both have been linked to early-onset parkinsonism.
Methods: We describe two new cases of SPG15 and investigate cellular changes in SPG15 and SPG11 patient-derived fibroblasts, seeking to identify shared pathogenic themes. Cells were evaluated for any abnormalities in cell division, DNA repair, endoplasmic reticulum, endosomes, and lysosomes.
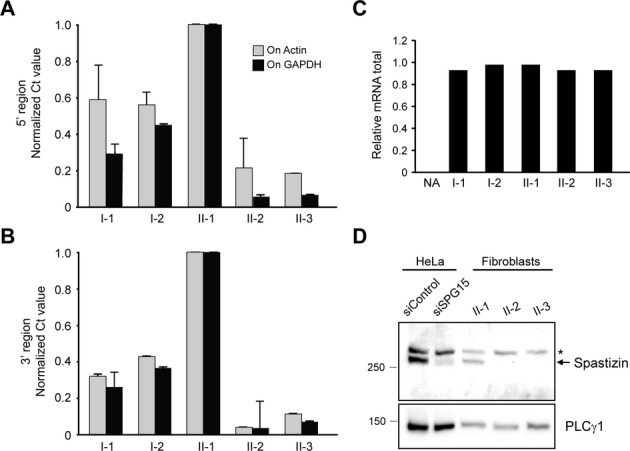
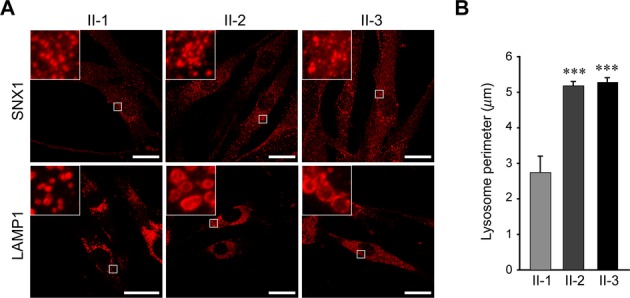
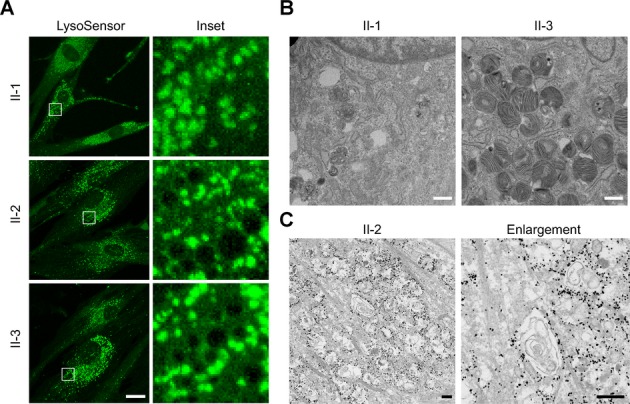
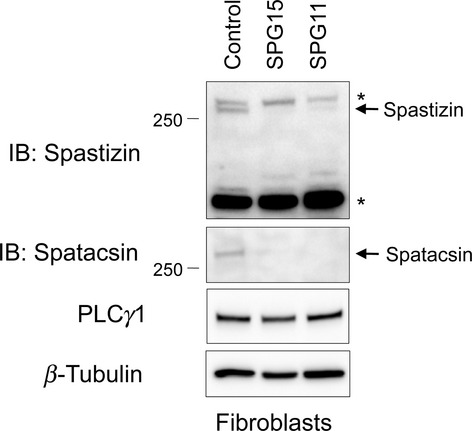
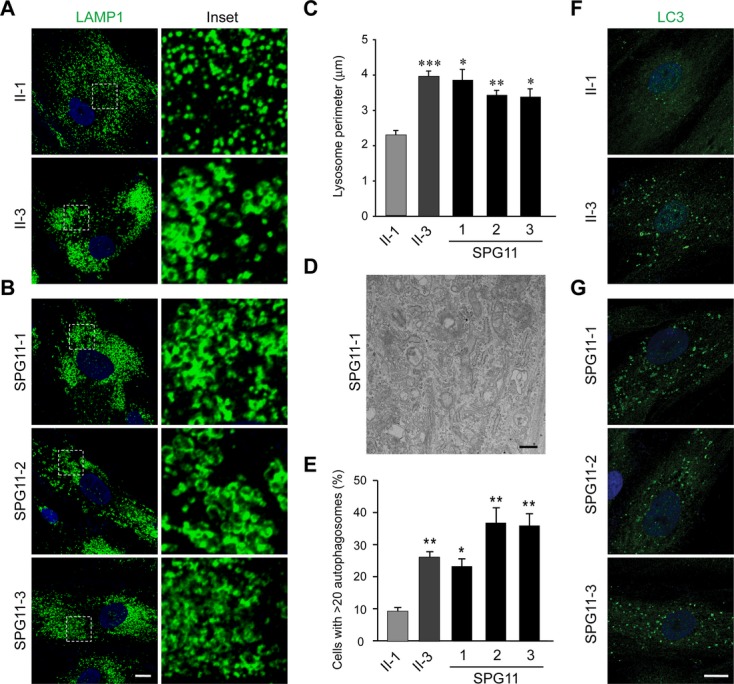
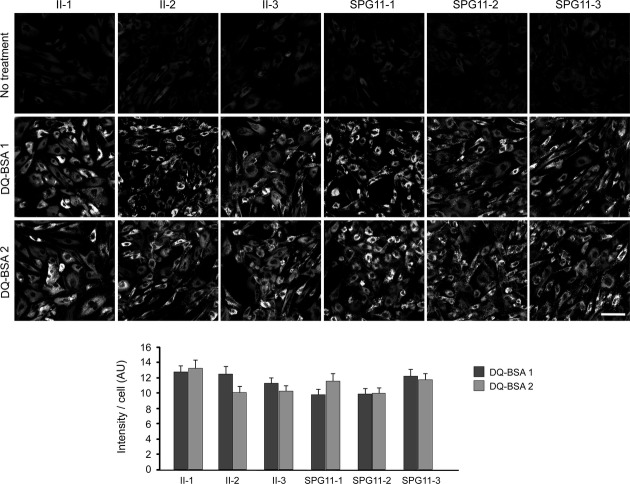
Results: Fibroblasts prepared from patients with SPG15 have selective enlargement of LAMP1-positive structures, and they consistently exhibited abnormal lysosomal storage by electron microscopy. A similar enlargement of LAMP1-positive structures was also observed in cells from multiple SPG11 patients, though prominent abnormal lysosomal storage was not evident. The stabilities of the SPG15 protein spastizin/ZFYVE26 and the SPG11 protein spatacsin were interdependent.
Interpretation: Emerging studies implicating these two proteins in interactions with the late endosomal/lysosomal adaptor protein complex AP-5 are consistent with shared abnormalities in lysosomes, supporting a converging mechanism for these two disorders. Recent work with Zfyve26-/- mice revealed a similar phenotype to human SPG15, and cells in these mice had endolysosomal abnormalities. SPG15 and SPG11 are particularly notable among HSPs because they can also present with juvenile parkinsonism, and this lysosomal trafficking or storage defect may be relevant for other forms of parkinsonism associated with lysosomal dysfunction.
Figures
References
-
- Finsterer J, Löscher W, Quasthoff S, et al. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. 2012;318:1–18. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
