

Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs
- PMID: 24999918
- PMCID: PMC4291073
- DOI: 10.1038/ncomms5330
Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs
Abstract
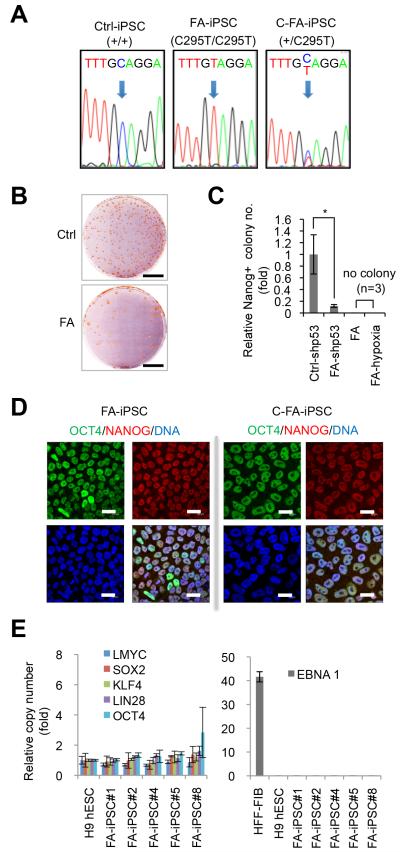
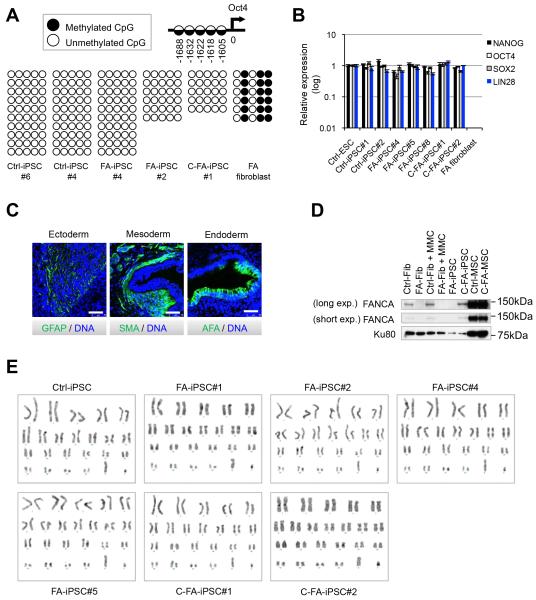
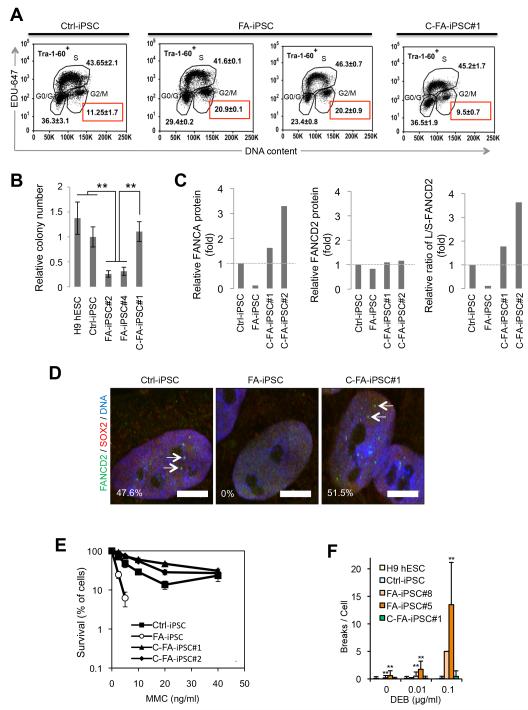
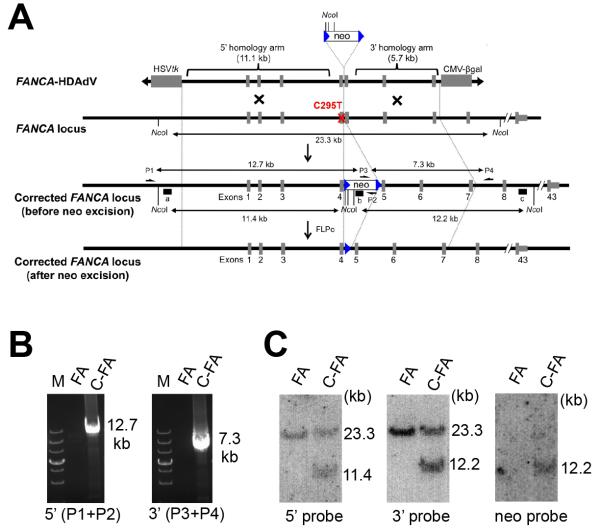
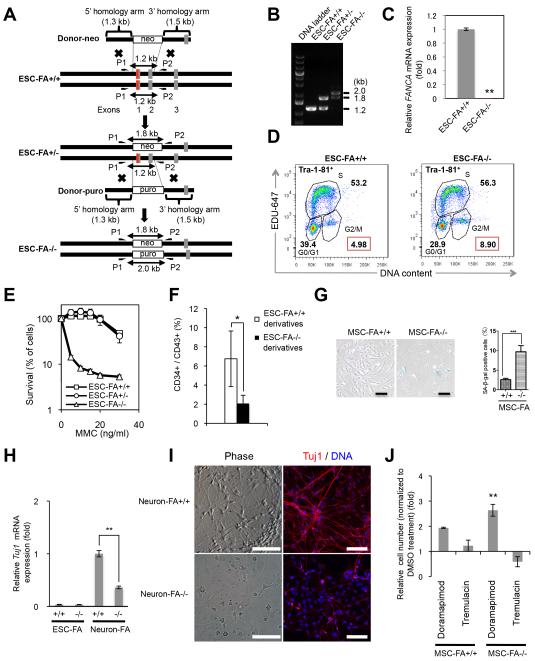
Fanconi anaemia (FA) is a recessive disorder characterized by genomic instability, congenital abnormalities, cancer predisposition and bone marrow (BM) failure. However, the pathogenesis of FA is not fully understood partly due to the limitations of current disease models. Here, we derive integration free-induced pluripotent stem cells (iPSCs) from an FA patient without genetic complementation and report in situ gene correction in FA-iPSCs as well as the generation of isogenic FANCA-deficient human embryonic stem cell (ESC) lines. FA cellular phenotypes are recapitulated in iPSCs/ESCs and their adult stem/progenitor cell derivatives. By using isogenic pathogenic mutation-free controls as well as cellular and genomic tools, our model serves to facilitate the discovery of novel disease features. We validate our model as a drug-screening platform by identifying several compounds that improve hematopoietic differentiation of FA-iPSCs. These compounds are also able to rescue the hematopoietic phenotype of FA patient BM cells.
Figures
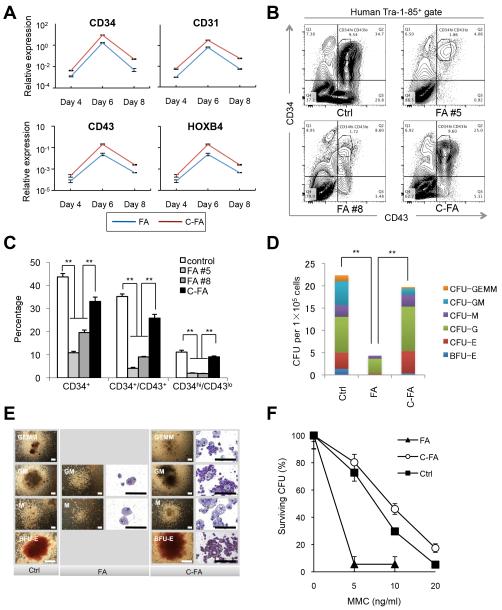
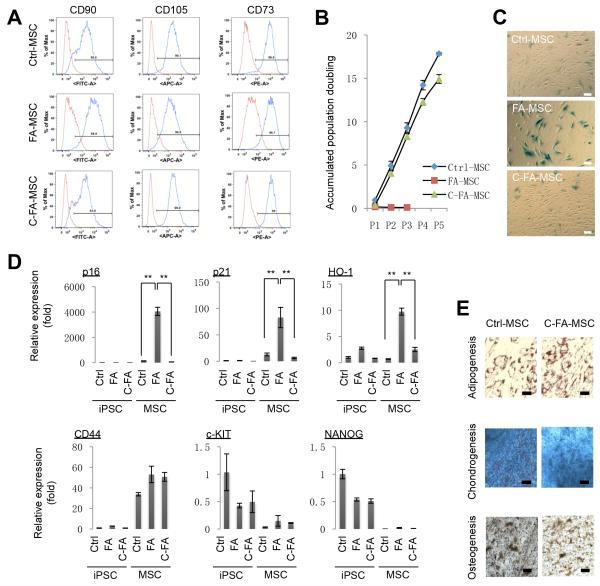
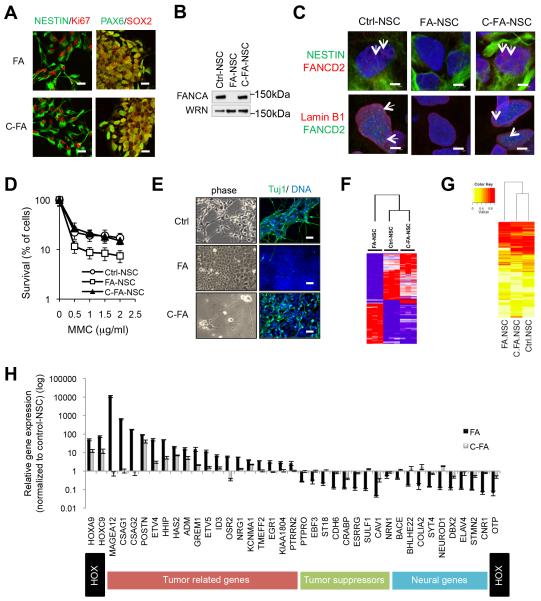
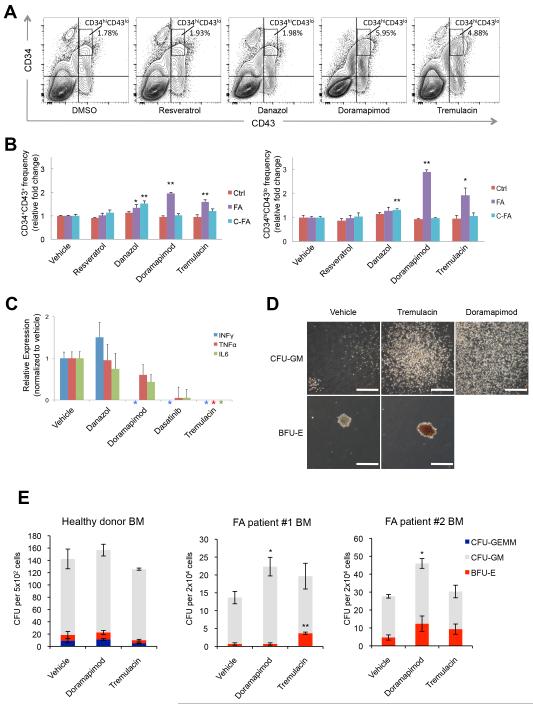
References
-
- Zhang QS, et al. Tempol protects against oxidative damage and delays epithelial tumor onset in Fanconi anemia mice. Cancer Res. 2008;68:1601–1608. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
