

Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome
- PMID: 25001218
- PMCID: PMC4154129
- DOI: 10.15252/emmm.201404044
Disruption of Mbd5 in mice causes neuronal functional deficits and neurobehavioral abnormalities consistent with 2q23.1 microdeletion syndrome
Abstract
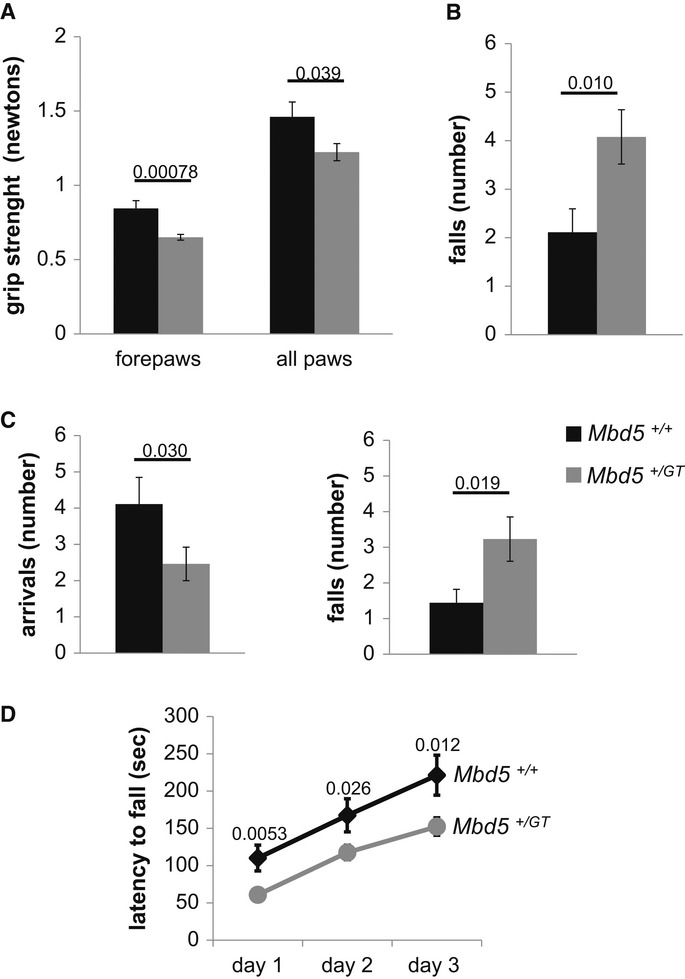
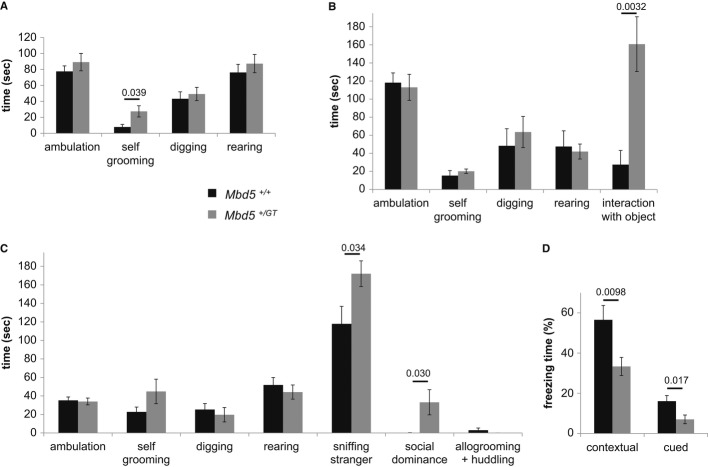
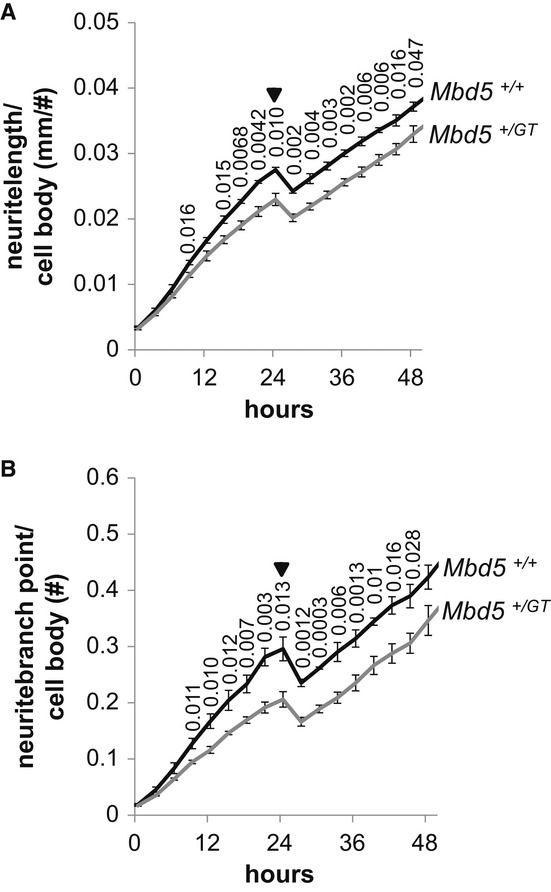
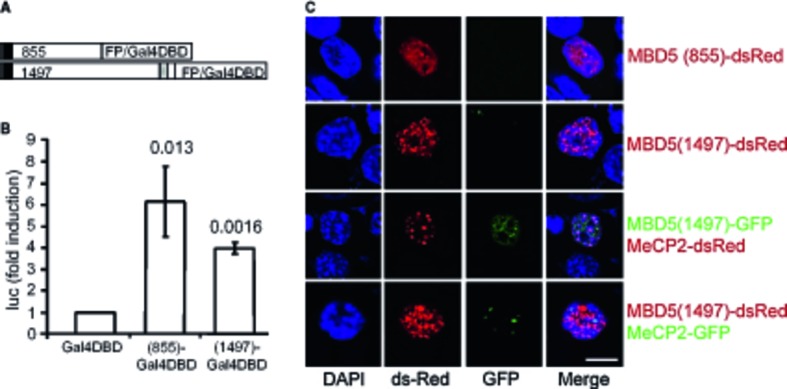
2q23.1 microdeletion syndrome is characterized by intellectual disability, motor delay, autistic-like behaviors, and a distinctive craniofacial phenotype. All patients carry a partial or total deletion of methyl-CpG-binding domain protein 5 (MBD5), suggesting that haploinsufficiency of this gene is responsible for the phenotype. To confirm this hypothesis and to examine the role of MBD5 in vivo, we have generated and characterized an Mbd5 gene-trap mouse model. Our study indicates that the Mbd5(+/) (GT) mouse model recapitulates most of the hallmark phenotypes observed in 2q23.1 deletion carriers including abnormal social behavior, cognitive impairment, and motor and craniofacial abnormalities. In addition, neuronal cultures uncovered a deficiency in neurite outgrowth. These findings support a causal role of MBD5 in 2q23.1 microdeletion syndrome and suggest a role for MBD5 in neuronal processes. The Mbd5(+/) (GT) mouse model will advance our understanding of the abnormal brain development underlying the emergence of 2q23.1 deletion-associated behavioral and cognitive symptoms.
Keywords: MBD5; autistic disorder; intellectual disability; mouse model.
© 2014 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
Comment in
-
Trapping MBD5 to understand 2q23.1 microdeletion syndrome.EMBO Mol Med. 2014 Aug;6(8):993-4. doi: 10.15252/emmm.201404324. EMBO Mol Med. 2014. PMID: 25001217 Free PMC article.
References
-
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) Washington, DC: American Psychiatric Press, Inc; 2000. Text Revision.
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. - PubMed
-
- Araki M, Nakahara M, Muta M, Itou M, Yanai C, Yamazoe F, Miyake M, Morita A, Araki M, Okamoto Y, et al. Database for exchangeable gene trap clones: pathway and gene ontology analysis of exchangeable gene trap clone mouse lines. Dev Growth Differ. 2014;56:161–174. - PubMed
-
- Bestor TH, Bourc'his D. Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb Symp Quant Biol. 2004;69:381–387. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
