

Expanding the computational toolbox for mining cancer genomes
- PMID: 25001846
- PMCID: PMC4168012
- DOI: 10.1038/nrg3767
Expanding the computational toolbox for mining cancer genomes
Abstract
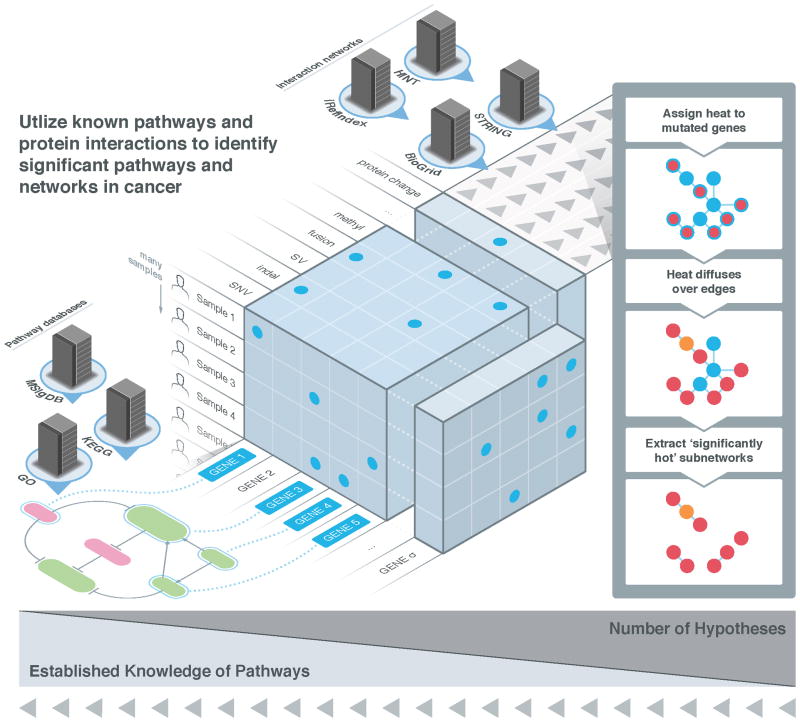
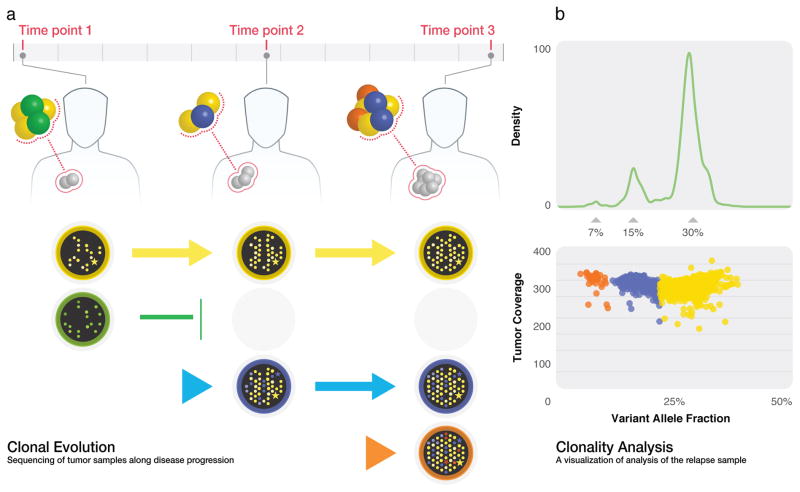
High-throughput DNA sequencing has revolutionized the study of cancer genomics with numerous discoveries that are relevant to cancer diagnosis and treatment. The latest sequencing and analysis methods have successfully identified somatic alterations, including single-nucleotide variants, insertions and deletions, copy-number aberrations, structural variants and gene fusions. Additional computational techniques have proved useful for defining the mutations, genes and molecular networks that drive diverse cancer phenotypes and that determine clonal architectures in tumour samples. Collectively, these tools have advanced the study of genomic, transcriptomic and epigenomic alterations in cancer, and their association to clinical properties. Here, we review cancer genomics software and the insights that have been gained from their application.
Figures
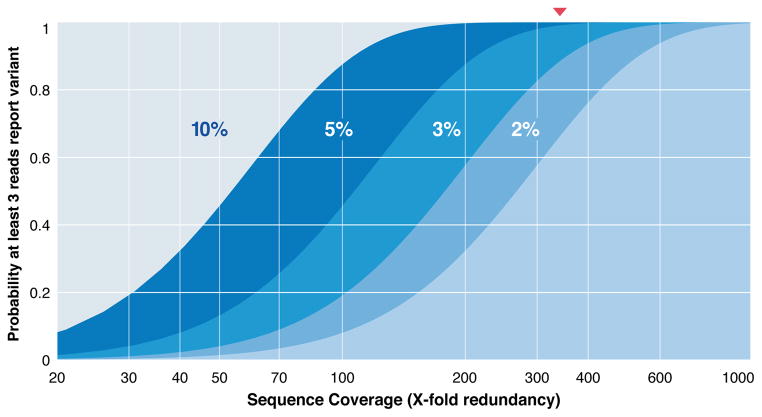
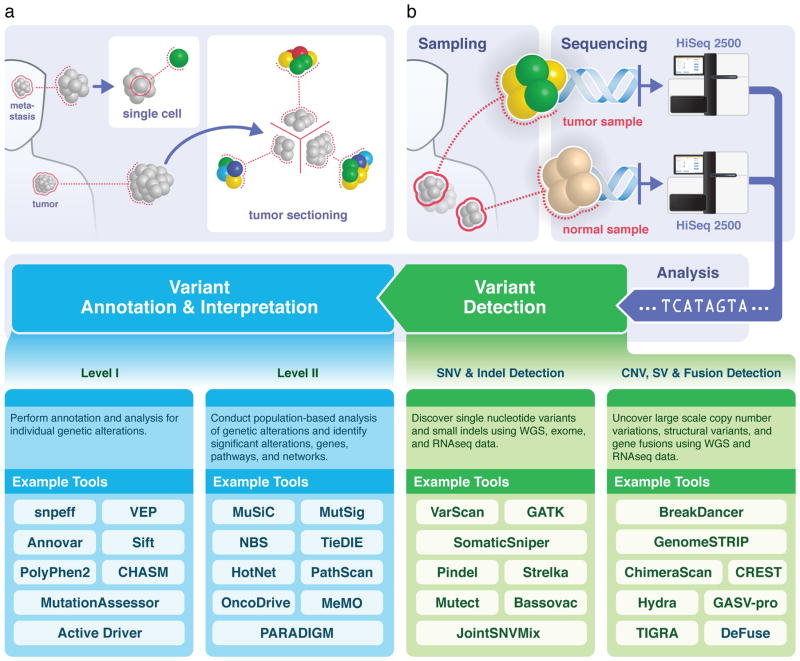
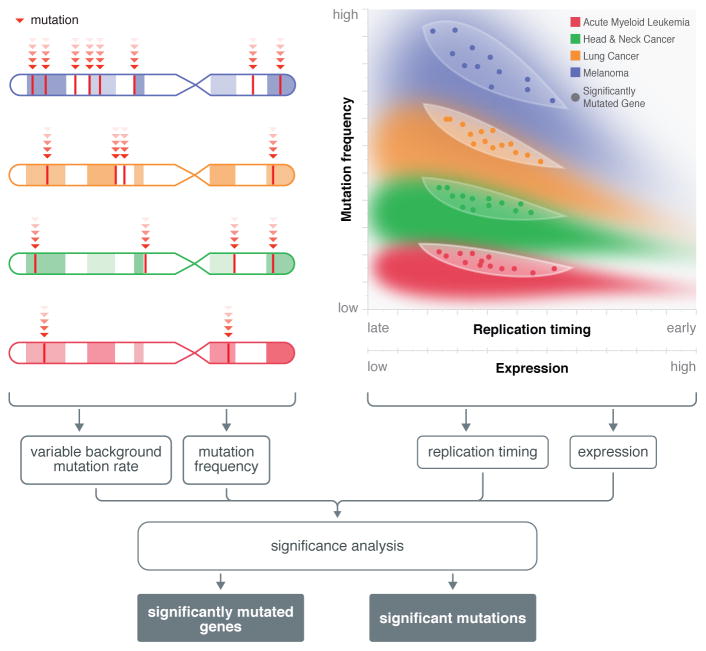
References
-
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. 1977. Biotechnology. 1992;24:104–108. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
