

Regulation of virulence of Entamoeba histolytica
- PMID: 25002094
- PMCID: PMC9006484
- DOI: 10.1146/annurev-micro-091313-103550
Regulation of virulence of Entamoeba histolytica
Abstract
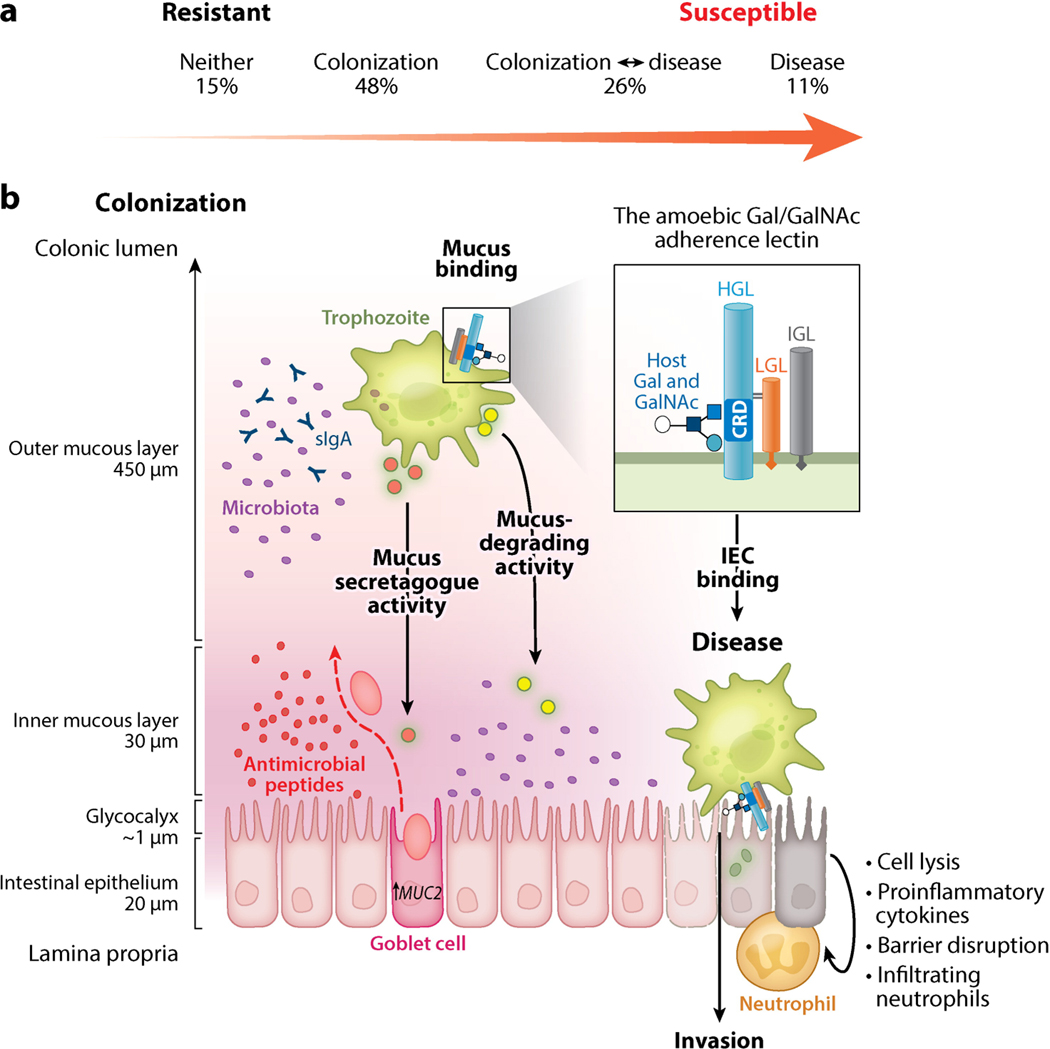
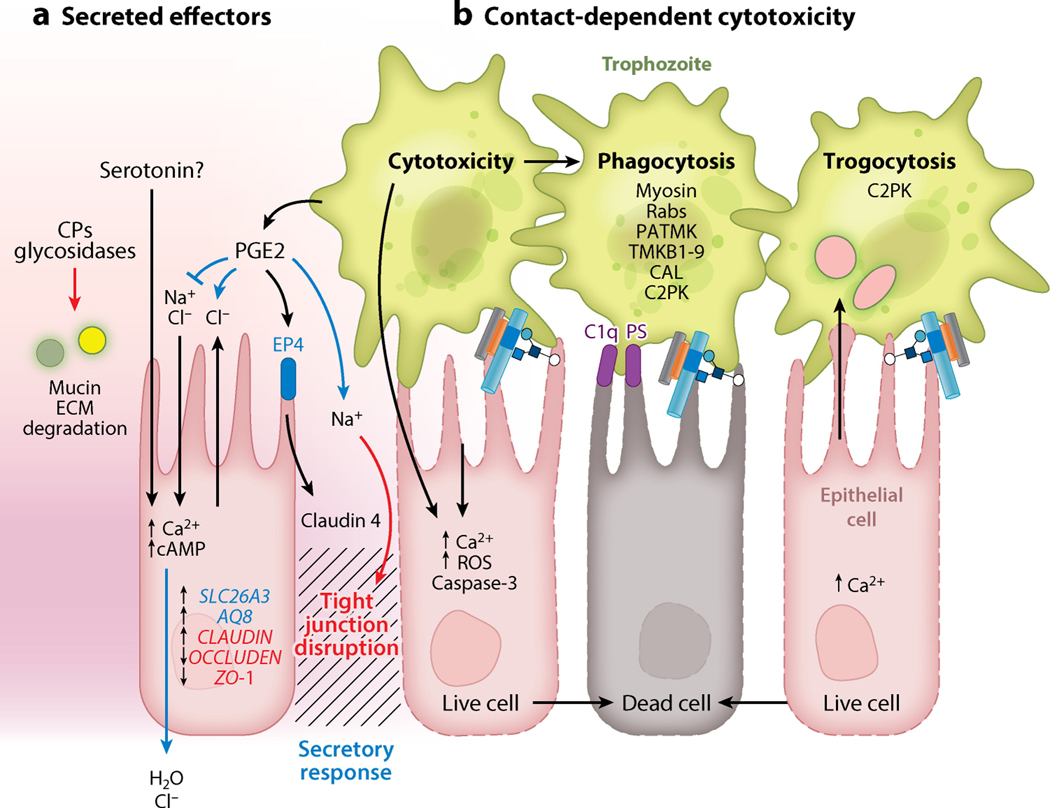
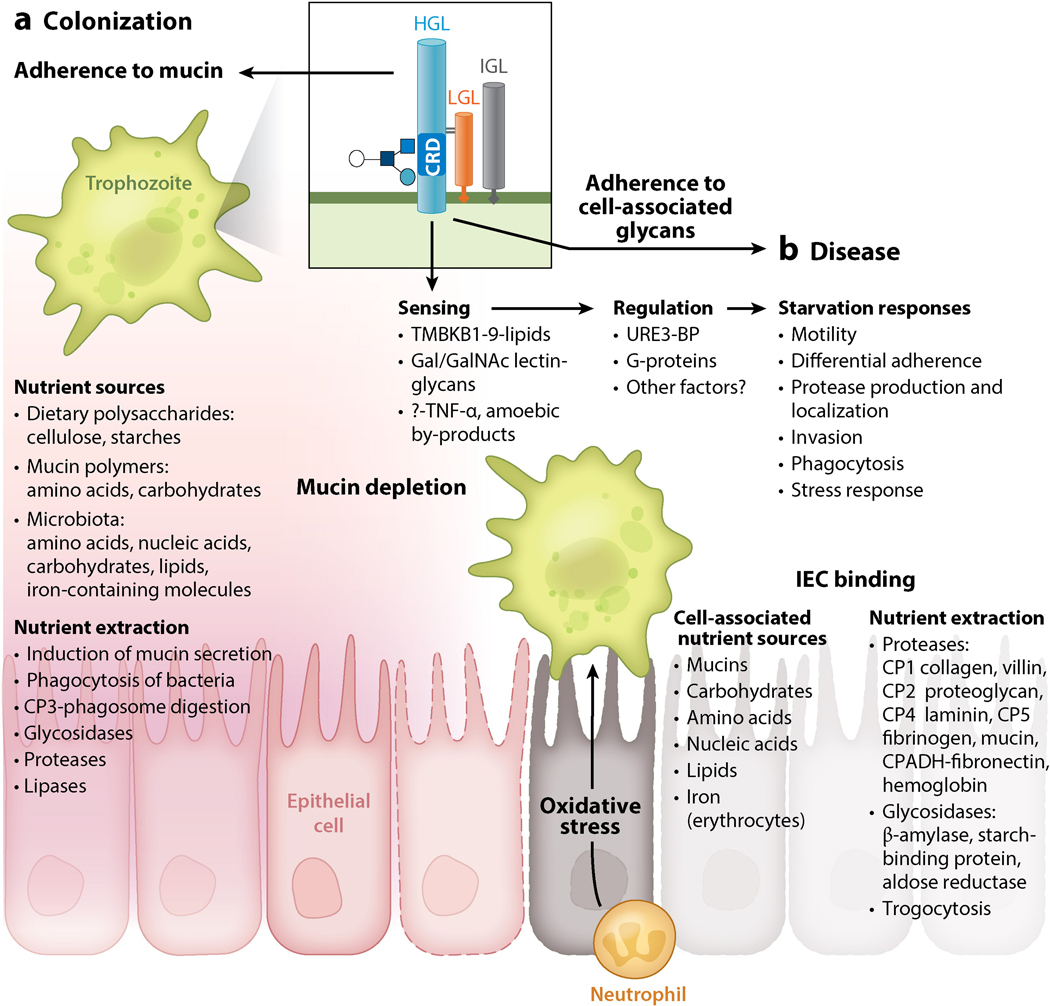
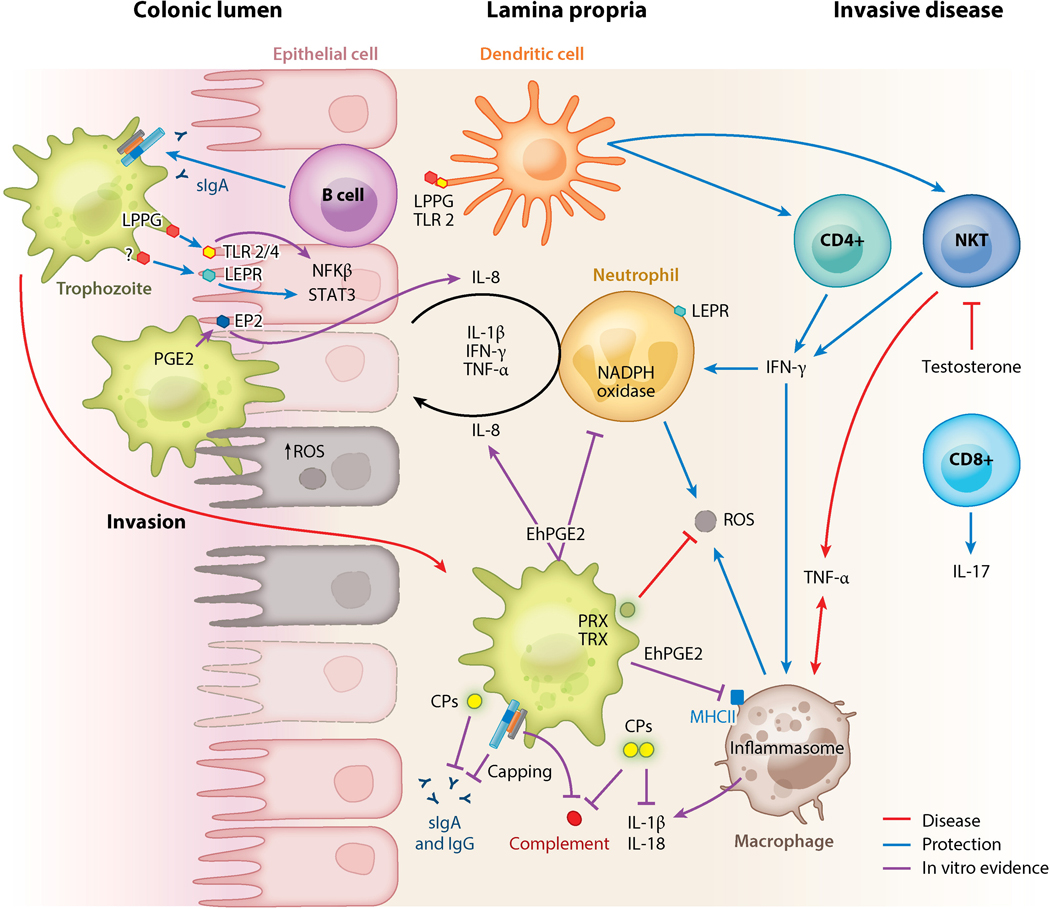
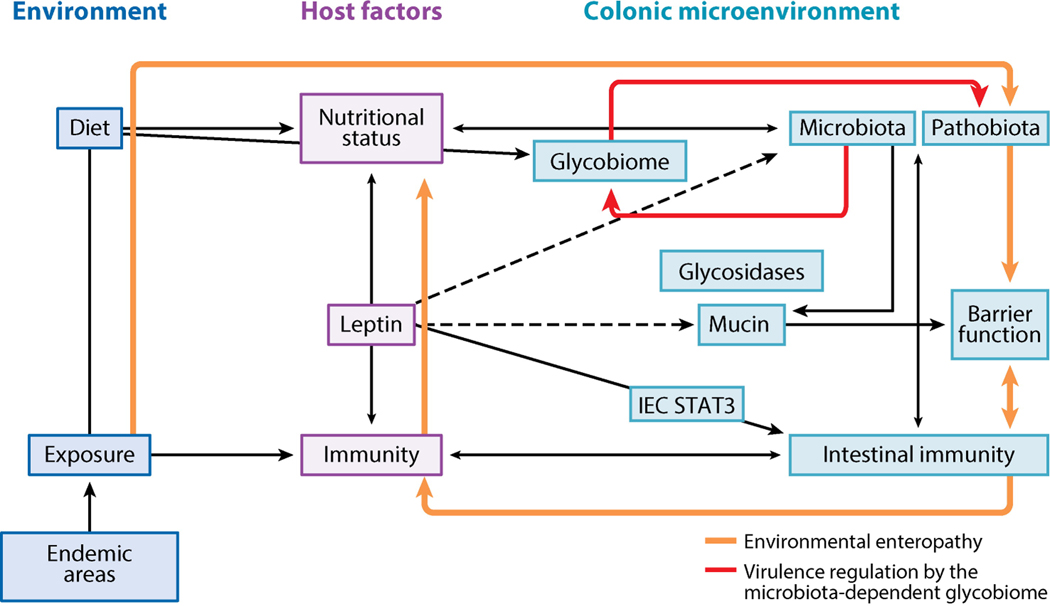
Entamoeba histolytica is the third-leading cause of parasitic mortality globally. E. histolytica infection generally does not cause symptoms, but the parasite has potent pathogenic potential. The origins, benefits, and triggers of amoebic virulence are complex. Amoebic pathogenesis entails depletion of the host mucosal barrier, adherence to the colonic lumen, cytotoxicity, and invasion of the colonic epithelium. Parasite damage results in colitis and, in some cases, disseminated disease. Both host and parasite genotypes influence the development of disease, as do the regulatory responses they govern at the host-pathogen interface. Host environmental factors determine parasite transmission and shape the colonic microenvironment E. histolytica infects. Here we highlight research that illuminates novel links between host, parasite, and environmental factors in the regulation of E. histolytica virulence.
Keywords: carbohydrate utilization; microbiota; mucus; pathobiota; virulence.
Figures
References
-
- Ackers J, Clark G, Diamond L, Duchene M, Cantellano M, et al. 1997. Who/paho/unesco report of a consultation of experts on amoebiasis
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
