

GNE myopathy: current update and future therapy
- PMID: 25002140
- PMCID: PMC4394625
- DOI: 10.1136/jnnp-2013-307051
GNE myopathy: current update and future therapy
Abstract
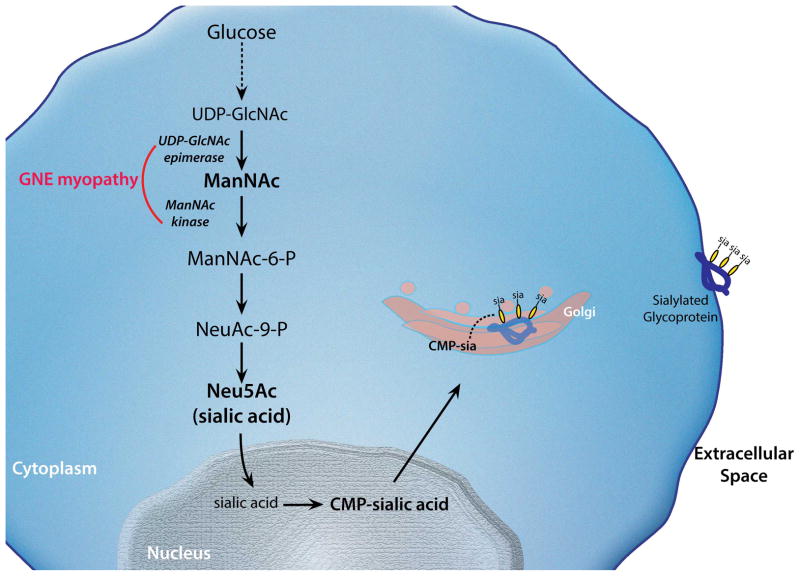
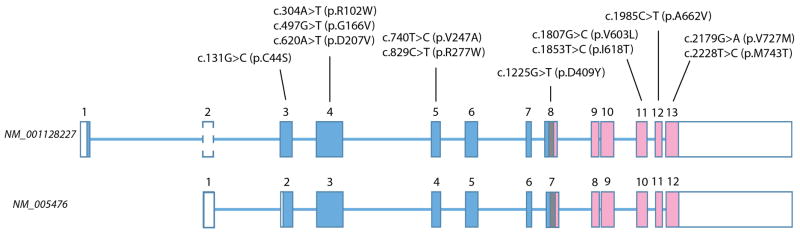
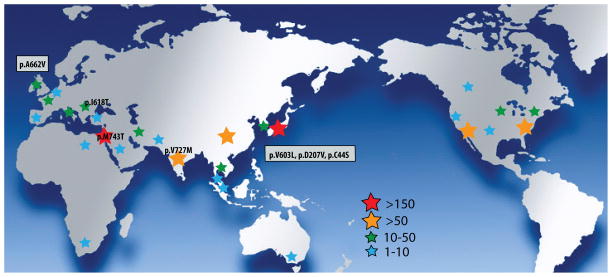
GNE myopathy is an autosomal recessive muscle disease caused by biallelic mutations in GNE, a gene encoding for a single protein with key enzymatic activities, UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase, in sialic acid biosynthetic pathway. The diagnosis should be considered primarily in patients presenting with distal weakness (foot drop) in early adulthood (other onset symptoms are possible too). The disease slowly progresses to involve other lower and upper extremities' muscles, with marked sparing of the quadriceps. Characteristic findings on biopsies of affected muscles include 'rimmed' (autophagic) vacuoles, aggregation of various proteins and fibre size variation. The diagnosis is confirmed by sequencing of the GNE gene. Note that we use a new mutation nomenclature based on the longest transcript (GenBank: NM_001128227), which encodes a 31-amino acid longer protein than the originally described one (GenBank: NM_005476), which has been used previously in most papers. Based upon the pathophysiology of the disease, recent clinical trials as well as early gene therapy trials have evaluated the use of sialic acid or N-acetylmannosamine (a precursor of sialic acid) in patients with GNE myopathy. Now that therapies are under investigation, it is critical that a timely and accurate diagnosis is made in patients with GNE myopathy.
Keywords: MUSCLE DISEASE; MYOPATHY; NEUROMUSCULAR.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://group.bmj.com/group/rights-licensing/permissions.
Conflict of interest statement
Dr. Argov is a co Pi and consultant for Ultragenyx. Dr. Nishino is a consultant for Ultragenyx.
Figures
References
-
- Nishino I, Noguchi S, Murayama K, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59(11):1689–93. - PubMed
-
- Argov Z, Eisenberg I, Grabov-Nardini G, et al. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. 2003;60(9):1519–23. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources