

Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3
- PMID: 25002506
- PMCID: PMC4115529
- DOI: 10.1073/pnas.1405601111
Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3
Abstract
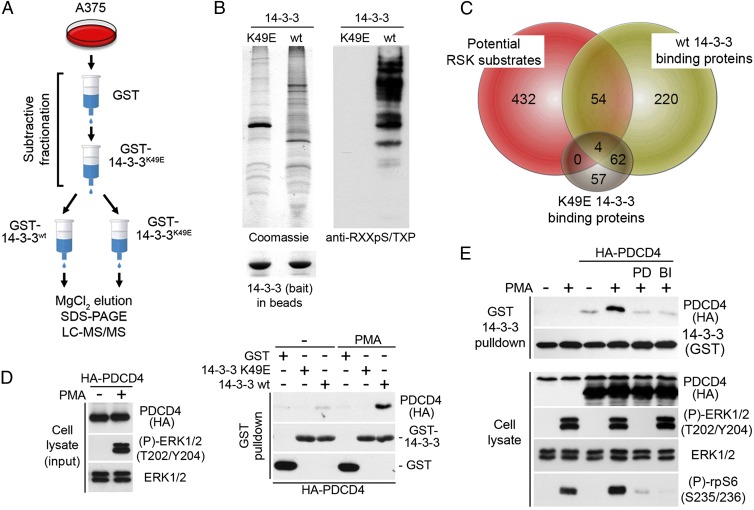
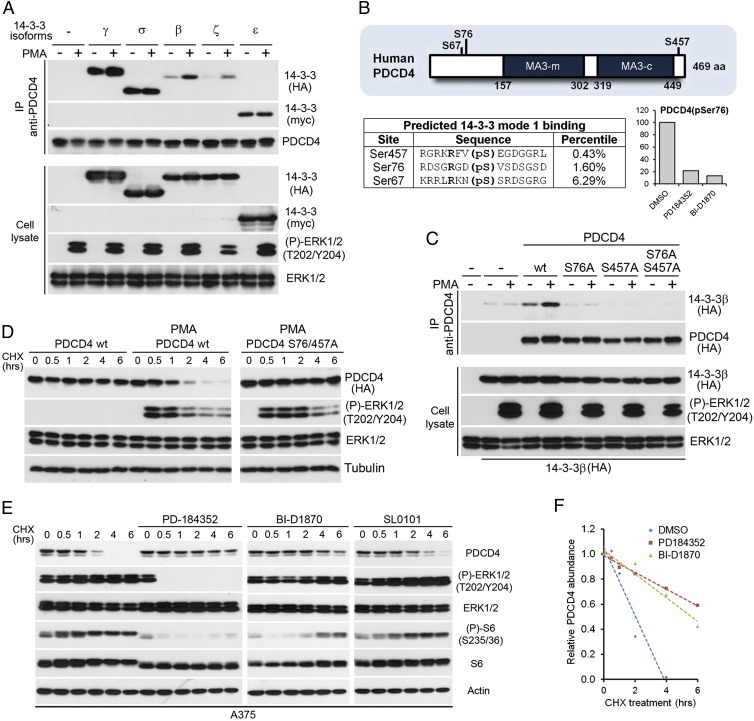
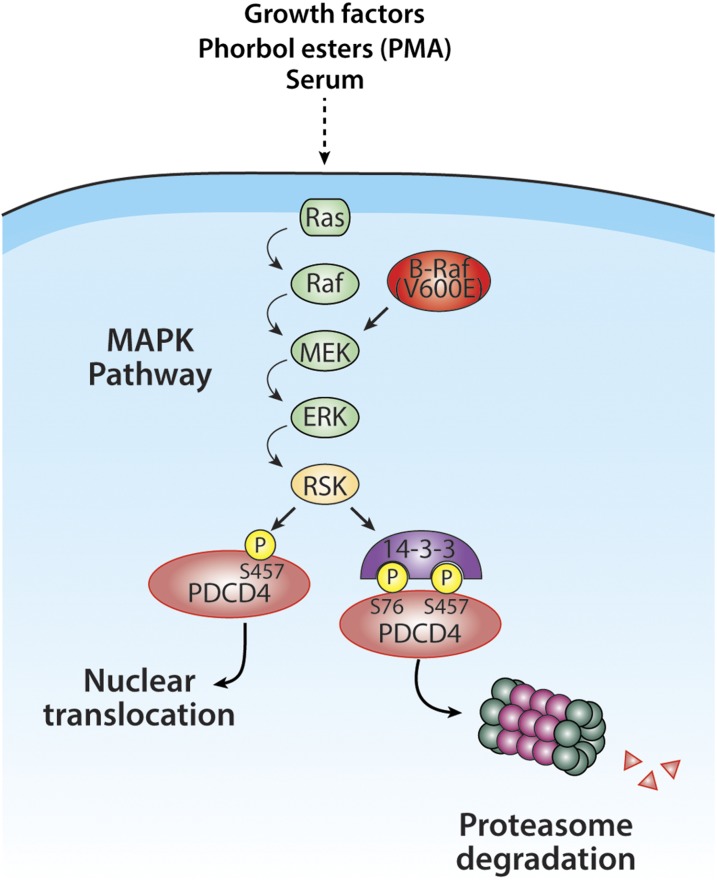
The Ras/MAPK signaling cascade regulates various biological functions, including cell growth and proliferation. As such, this pathway is frequently deregulated in several types of cancer, including most cases of melanoma. RSK (p90 ribosomal S6 kinase) is a MAPK-activated protein kinase required for melanoma growth and proliferation, but relatively little is known about its exact function and the nature of its substrates. Herein, we used a quantitative phosphoproteomics approach to define the signaling networks regulated by RSK in melanoma. To more accurately predict direct phosphorylation substrates, we defined the RSK consensus phosphorylation motif and found significant overlap with the binding consensus of 14-3-3 proteins. We thus characterized the phospho-dependent 14-3-3 interactome in melanoma cells and found that a large proportion of 14-3-3 binding proteins are also potential RSK substrates. Our results show that RSK phosphorylates the tumor suppressor PDCD4 (programmed cell death protein 4) on two serine residues (Ser76 and Ser457) that regulate its subcellular localization and interaction with 14-3-3 proteins. We found that 14-3-3 binding promotes PDCD4 degradation, suggesting an important role for RSK in the inactivation of PDCD4 in melanoma. In addition to this tumor suppressor, our results suggest the involvement of RSK in a vast array of unexplored biological functions with relevance in oncogenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
RSK-mediated down-regulation of PDCD4 is required for proliferation, survival, and migration in a model of triple-negative breast cancer.Oncotarget. 2016 May 10;7(19):27567-83. doi: 10.18632/oncotarget.8375. Oncotarget. 2016. PMID: 27028868 Free PMC article.
-
Proteomic Analysis Reveals a Role for RSK in p120-catenin Phosphorylation and Melanoma Cell-Cell Adhesion.Mol Cell Proteomics. 2020 Jan;19(1):50-64. doi: 10.1074/mcp.RA119.001811. Epub 2019 Nov 2. Mol Cell Proteomics. 2020. PMID: 31678930 Free PMC article.
-
Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase.Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13489-94. doi: 10.1073/pnas.0405659101. Epub 2004 Sep 1. Proc Natl Acad Sci U S A. 2004. PMID: 15342917 Free PMC article.
-
The RSK factors of activating the Ras/MAPK signaling cascade.Front Biosci. 2008 May 1;13:4258-75. doi: 10.2741/3003. Front Biosci. 2008. PMID: 18508509 Review.
-
The RSK family of kinases: emerging roles in cellular signalling.Nat Rev Mol Cell Biol. 2008 Oct;9(10):747-58. doi: 10.1038/nrm2509. Nat Rev Mol Cell Biol. 2008. PMID: 18813292 Review.
Cited by
-
Recent findings and technological advances in phosphoproteomics for cells and tissues.Expert Rev Proteomics. 2015;12(5):469-87. doi: 10.1586/14789450.2015.1078730. Expert Rev Proteomics. 2015. PMID: 26400465 Free PMC article. Review.
-
KSR1 and EPHB4 Regulate Myc and PGC1β To Promote Survival of Human Colon Tumors.Mol Cell Biol. 2016 Aug 12;36(17):2246-61. doi: 10.1128/MCB.00087-16. Print 2016 Sep 1. Mol Cell Biol. 2016. PMID: 27273865 Free PMC article.
-
Phosphorylation and Signal Transduction Pathways in Translational Control.Cold Spring Harb Perspect Biol. 2019 Jul 1;11(7):a033050. doi: 10.1101/cshperspect.a033050. Cold Spring Harb Perspect Biol. 2019. PMID: 29959191 Free PMC article. Review.
-
ERK signalling: a master regulator of cell behaviour, life and fate.Nat Rev Mol Cell Biol. 2020 Oct;21(10):607-632. doi: 10.1038/s41580-020-0255-7. Epub 2020 Jun 23. Nat Rev Mol Cell Biol. 2020. PMID: 32576977 Review.
-
PLEKHA5 regulates tumor growth in metastatic melanoma.Cancer. 2020 Mar 1;126(5):1016-1030. doi: 10.1002/cncr.32611. Epub 2019 Nov 26. Cancer. 2020. PMID: 31769872 Free PMC article.
References
-
- Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–3239. - PubMed
-
- Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. - PubMed
-
- Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008;27(7):877–895. - PubMed
-
- Fecher LA, Amaravadi RK, Flaherty KT. The MAPK pathway in melanoma. Curr Opin Oncol. 2008;20(2):183–189. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
