

High-throughput targeted repeat element bisulfite sequencing (HT-TREBS): genome-wide DNA methylation analysis of IAP LTR retrotransposon
- PMID: 25003790
- PMCID: PMC4086960
- DOI: 10.1371/journal.pone.0101683
High-throughput targeted repeat element bisulfite sequencing (HT-TREBS): genome-wide DNA methylation analysis of IAP LTR retrotransposon
Abstract
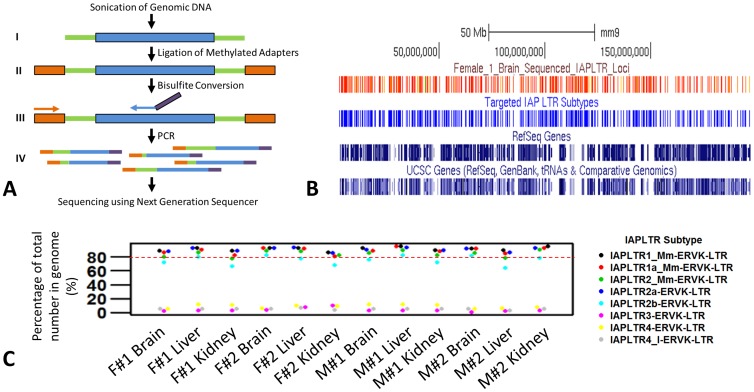
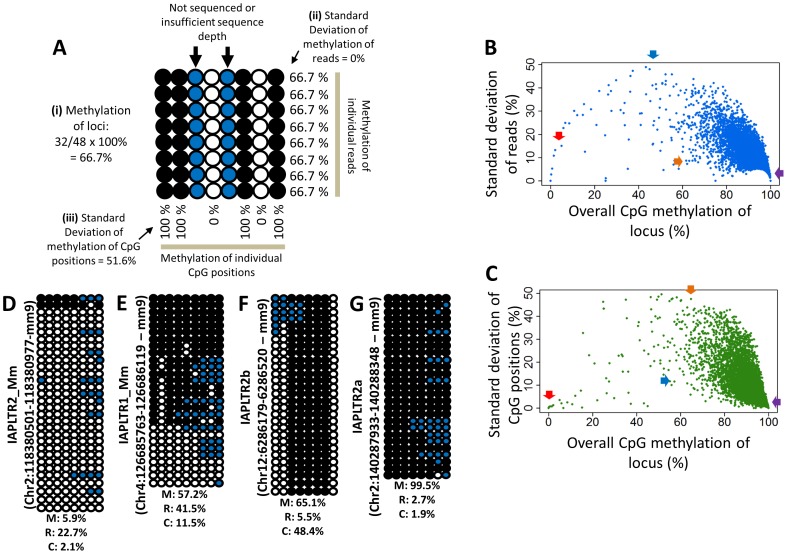
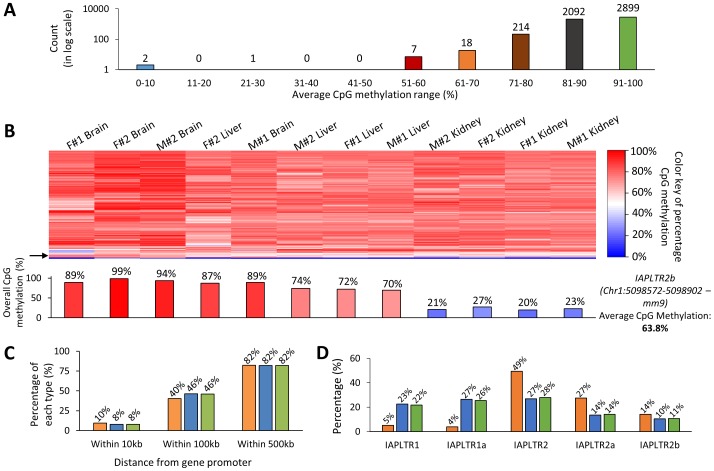
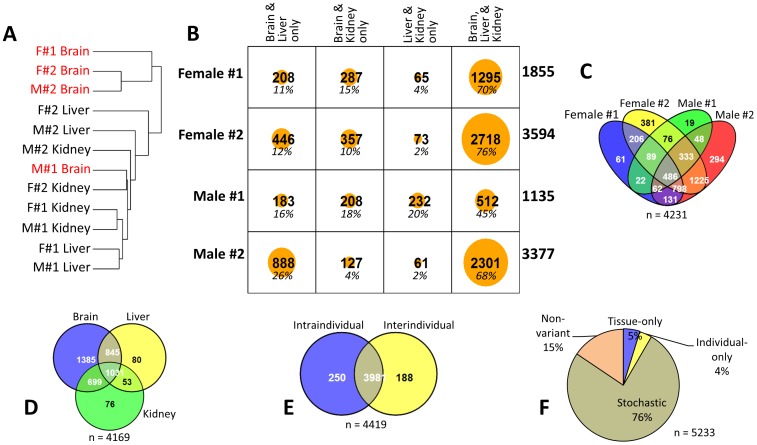
In vertebrates, DNA methylation-mediated repression of retrotransposons is essential for the maintenance of genomic integrity. In the current study, we developed a technique termed HT-TREBS (High-Throughput Targeted Repeat Element Bisulfite Sequencing). This technique is designed to measure the DNA methylation levels of individual loci of any repeat families with next-generation sequencing approaches. To test the feasibility of HT-TREBS, we analyzed the DNA methylation levels of the IAP LTR family using a set of 12 different genomic DNA isolated from the brain, liver and kidney of 4 one-week-old littermates of the mouse strain C57BL/6N. This technique has successfully generated the CpG methylation data of 5,233 loci common in all the samples, representing more than 80% of the individual loci of the five targeted subtypes of the IAP LTR family. According to the results, approximately 5% of the IAP LTR loci have less than 80% CpG methylation levels with no genomic position preference. Further analyses of the IAP LTR loci also revealed the presence of extensive DNA methylation variations between different tissues and individuals. Overall, these data demonstrate the efficiency and robustness of the new technique, HT-TREBS, and also provide new insights regarding the genome-wide DNA methylation patterns of the IAP LTR repeat elements.
Conflict of interest statement
Figures
Similar articles
-
Retrotransposon-based profiling of mammalian epigenomes: DNA methylation of IAP LTRs in embryonic stem, somatic and cancer cells.Genomics. 2014 Dec;104(6 Pt B):538-44. doi: 10.1016/j.ygeno.2014.09.009. Epub 2014 Sep 29. Genomics. 2014. PMID: 25277721 Free PMC article.
-
Locus-specific DNA methylation analysis of retrotransposons in ES, somatic and cancer cells using High-Throughput Targeted Repeat Element Bisulfite Sequencing.Genom Data. 2015 Mar 1;3:87-89. doi: 10.1016/j.gdata.2014.11.013. Genom Data. 2015. PMID: 25554740 Free PMC article.
-
High-Throughput Targeted Repeat Element Bisulfite Sequencing (HT-TREBS).Methods Mol Biol. 2019;1908:219-228. doi: 10.1007/978-1-4939-9004-7_15. Methods Mol Biol. 2019. PMID: 30649731
-
Tools and Strategies for Analysis of Genome-Wide and Gene-Specific DNA Methylation Patterns.Methods Mol Biol. 2017;1537:249-277. doi: 10.1007/978-1-4939-6685-1_15. Methods Mol Biol. 2017. PMID: 27924599 Review.
-
Analyzing the cancer methylome through targeted bisulfite sequencing.Cancer Lett. 2013 Nov 1;340(2):171-8. doi: 10.1016/j.canlet.2012.10.040. Epub 2012 Nov 28. Cancer Lett. 2013. PMID: 23200671 Free PMC article. Review.
Cited by
-
Retrotransposon-based profiling of mammalian epigenomes: DNA methylation of IAP LTRs in embryonic stem, somatic and cancer cells.Genomics. 2014 Dec;104(6 Pt B):538-44. doi: 10.1016/j.ygeno.2014.09.009. Epub 2014 Sep 29. Genomics. 2014. PMID: 25277721 Free PMC article.
-
Performances of Different Fragment Sizes for Reduced Representation Bisulfite Sequencing in Pigs.Biol Proced Online. 2017 Jun 7;19:5. doi: 10.1186/s12575-017-0054-5. eCollection 2017. Biol Proced Online. 2017. PMID: 28596713 Free PMC article.
-
A novel quantitative trait locus implicates Msh3 in the propensity for genome-wide short tandem repeat expansions in mice.Genome Res. 2023 May;33(5):689-702. doi: 10.1101/gr.277576.122. Epub 2023 May 1. Genome Res. 2023. PMID: 37127331 Free PMC article.
-
Integrated small copy number variations and epigenome maps of disorders of sex development.Hum Genome Var. 2016 Jun 9;3:16012. doi: 10.1038/hgv.2016.12. eCollection 2016. Hum Genome Var. 2016. PMID: 27340555 Free PMC article.
-
Locus-specific DNA methylation analysis of retrotransposons in ES, somatic and cancer cells using High-Throughput Targeted Repeat Element Bisulfite Sequencing.Genom Data. 2015 Mar 1;3:87-89. doi: 10.1016/j.gdata.2014.11.013. Genom Data. 2015. PMID: 25554740 Free PMC article.
References
-
- Schmid CW, Deininger PL (1975) Sequence organization of the human genome. Cell 6: 345–358. - PubMed
-
- Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nature Rev Genet 3: 370–379. - PubMed
-
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
